Received: December 2025
DOI 10.17677/fn20714807.2026.01.01
Fluorine Notes, 2026, 164, 1-2
A SERIES OF LECTURES. SOME DIRECTIONS OF FLUORINE CHEMISTRY
COMMUNICATION 2. PART 3. FLUORINATION OF ALIPHATIC COMPOUNDS WITH HIGH VALENCY METAL FLUORIDES IN THE WORKS OF RUSSIAN RESEARCHERS
V.V. Kornilov
Abstract: The results of fluorination of methane, acetylene, ethylene, and a number of polyfluoroethanes with high valency metal fluorides (HVMFs) are reviewed. Possible mechanisms of reactions of C1-C2 hydrocarbons and C1-C2 polyfluoroalkanes with HVMFs, as well as rearrangements associated with H-migration at intermediate steps of fluorination, are considered. The effect of hydrogen fluoride as a catalyst on fluorination processes using HVMFs is also considered. The hypothesis about the possible influence of adsorption of organic molecules on the surface of HVMFs on the ratio of polyfluoroethanes isomers is discussed
Keywords: high valency metal fluorides (HVMFs), tetrafluoroethane isomers, cobalt trifluoride, manganese trifluoride, cerium tetrafluoride, carbocations, cation-radical mechanism, adsorption, complex fluorides.
Introduction
In the previous parts of the communication [1,2], a brief overview of the fluorination of organic compounds with high valency metal fluorides (HVMFs) was provided, and probable mechanisms of fluorination of aromatic and aliphatic compounds were considered based on publications by foreign authors. This article is disclosing the results of Russian researchers in the field of fluorination of aliphatic C1-C2 compounds and C1-C2 polyfluoroalkanes with HVMFs.
2. Fluorination of C1-C2 hydrocarbons and C1-C2 polyfluoroalkanes with HVMFs
The fluorination of lower hydrocarbons (methane, ethylene, etc.) and C1-C2 polyfluoroalkanes with HVMFs was studied in the 70s-90s of the 20th century at RSC “Applied Chemistry” (former State Institute of Applied Chemistry) (Saint-Petersburg). A significant part of the results was published in articles [3-9] and review [10].
Vertical reactors with an internal diameter of 18÷36 mm and a height of 45÷100 cm were used to study the processes, with the initial hydrocarbons fed into the bottom of the reactor. This design had a number of advantages for research compared to horizontal reactors equipped with a coaxial paddle stirrer, which were used by foreign authors [1,2].
Firstly, vertical reactors had a simple design and, accordingly, increased reliability.
Secondly, horizontal reactors with stirrers were filled with fluorides powder to 2/3÷5/6 of their volume, therefore they could have an empty space in the upper part of the reactor. The vertical reactors were completely filled with a fluorinating reagent, which ensured uniform flow of gaseous hydrocarbon through the entire HVMFs layer and excluded hydrocarbon breakthrough without fluorination even at short contact times (several seconds). It was experimentally established that the finely dispersed state and uniformity of the filling remained unchanged even after a large number of regeneration-synthesis cycles.
Thirdly, the vertical design used allowed for minimizing the inaccuracy in measuring the reactor temperature profile. For this purpose, the reactor was equipped with either several thin-walled pockets for thermocouples (every 10-12 cm in height) or a thin-walled tube (d=3 mm) located along the vertical reactor axis.
In some syntheses, to study the dependence of the product composition on the thickness of the HVMFs layer, the reactor was equipped with several samplers along the height.
It was found that during fluorination without dilution with an inert gas, the reaction proceeded in a narrow zone of the HVMF’s layer (2-5 cm) with a gradual shift of the reaction zone along the height of the reactor. The displacement of such a zone along the height of the reactor could be observed by the temperature profile. As the active reaction zone approached, a sharp increase in temperature was recorded, with the temperature value passing through a peak and then decreasing. The value of the temperature gradient depended on the rate of hydrocarbon supply and the possibility of heat removal from the reaction zone. If the peak temperature reached ≈250°C, then an almost complete transformation of cobalt trifluoride into difluoride occurred. This was determined as follows: the process was stopped when the temperature peaked approximately in the middle of the reactor, the reactor was blown with an inert gas, cooled, and disassembled. The reacted layer consisted of purple cobalt difluoride. The unreacted layer above the reaction zone consisted of almost pure brown cobalt trifluoride. Diluting the initial hydrocarbon with an inert gas allowed for studies to be conducted under isothermal conditions. In this case, the entire HVMF layer was the reaction zone.
2.1. Fluorination of methane with cobalt trifluoride
The results of methane (Fig.1), difluoromethane, and trifluoromethane fluorination with cobalt trifluoride were disclosed in [3]. More detailed fluorination results of difluoromethane (temperature range 184÷351ºC) and trifluoromethane (temperature range 210÷362ºC) were published in [8] somewhat later.
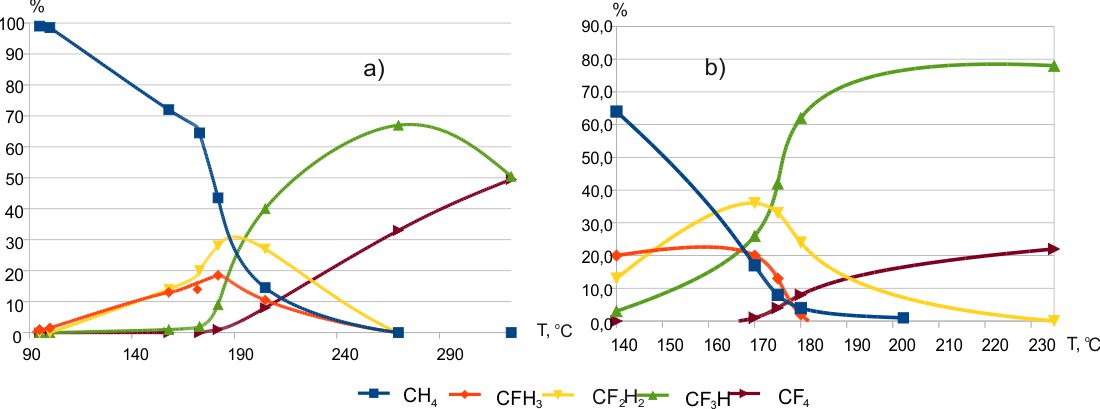
Fig. 1. Composition of methane fluorination products (а- mixture of 5% CH4 and 95% He, b- 100% CH4) [3].
By analogy with the fluorination of other hydrocarbons, it was proposed that the process proceeds via a cation-radical mechanism (Scheme 1) [3].

Scheme 1.
Obviously, the fluorination of methane cannot proceed through the formation of intermediate alkenes. Since no fundamental differences are visible between the process of methane fluorination and other hydrocarbons, it can be assumed that the thesis about the formation of olefins at the initial step of alkanes fluorination (for example, propane [11]) seems controversial (see the previous part of the communication [2]).
2.2. Fluorination of acetylene with cobalt trifluoride and cerium tetrafluoride
The results of acetylene fluorination with cobalt trifluoride and cerium tetrafluoride were presented in [4,5].
It was found that fluorination with cerium tetrafluoride began at temperatures above 100ºC (Fig.2). Tetrafluoroethanes (the Fig.2 shows the total content of 1,1,2,2- and 1,1,1,2-tetrafluoroethanes in a ratio of ≈4:1) and pentafluoroethane were formed over a wide temperature range.
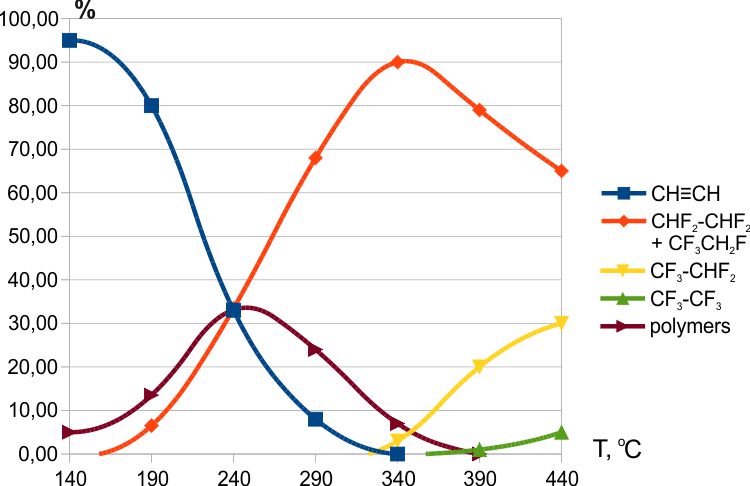
Fig. 2. Fluorination of acetylene with CeF4.
A characteristic feature of the interaction of acetylene with cerium tetrafluoride was the obtaining of liquid oligomeric compounds in the reaction products. A possible mechanism for the formation of such compounds will be discussed below (see 2.4.4).
The interaction of acetylene with CoF3 began at T≈ 80 °C, and at 250 °C more than 50% pentafluoroethane was obtained in the reaction products (Fig. 3) [5].
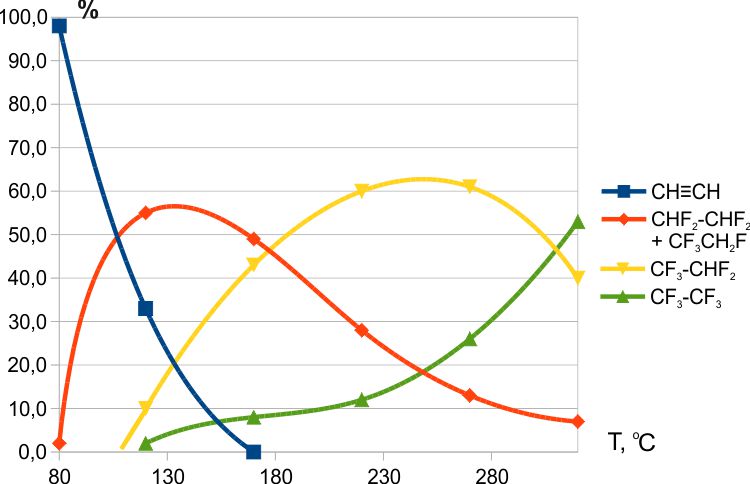
Fig. 3. Fluorination of acetylene with CoF3.
Fig. 3 shows the total content of two isomers of C2H2F4 with a ratio of 1,1,2,2-tetrafluoroethane to 1,1,1,2-tetrafluoroethane as ≈3:1. The possible mechanism of the obtaining of 1,1,1,2-tetrafluoroethane will be discussed below (see 2.4.3).
2.3. Fluorination of ethylene with cobalt trifluoride, manganese trifluoride and cerium tetrafluoride
In papers [4-6], the results of fluorination of ethylene and some polyfluoroalkanes with cobalt trifluoride, manganese trifluoride and cerium tetrafluoride were considered.
It was found that the reaction of ethylene with cobalt trifluoride began already at room temperature, and at 50ºC, traces of ethylene were absent in the fluorination products. At temperatures above 80-100 °C, small amounts of polyfluoromethanes could be produced (Fig. 4). The reaction of ethylene with manganese trifluoride began at ≈140ºC, and ethylene remained in the fluorination products up to 260-270ºC (Fig.5). The reaction of ethylene with cerium tetrafluoride began at ≈125ºC, and traces of ethylene remained in the fluorination products up to 235ºC (Fig.6). In addition, at low temperatures of fluorination with CeF4 the traces of fluoroethylene were detected.
The obtained results confirmed the thesis of the dependence of the power of the fluorinating agent on the oxidation potential of the metal cation. The beginning of interaction with weak cerium tetrafluoride at a lower temperature compared to stronger manganese trifluoride can be explained by a competing process occurring via the mechanism of ionic (radical) polymerization, which led to the formation of liquid oligomeric products (See 2.4.4).
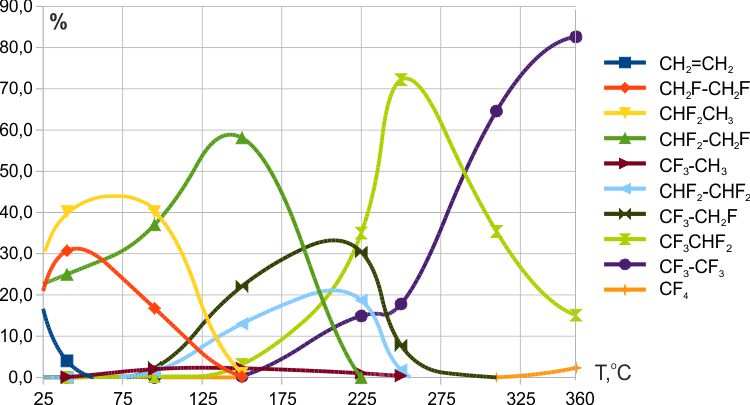
Fig. 4. Fluorination of ethylene with CoF3 (T= 25→360ºС, contact time ≈7 min.).
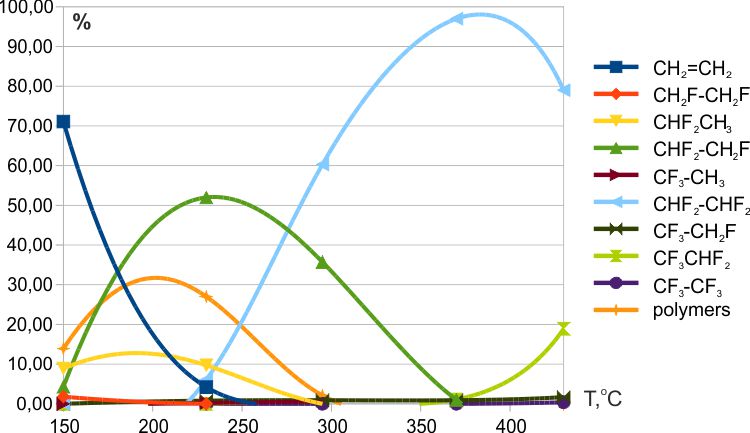
Fig.5. Fluorination of ethylene with CeF4 (T= 150→430º, contact time ≈7 min.).
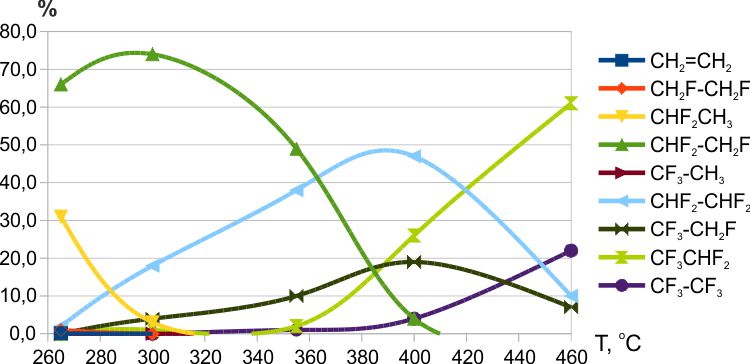
Fig.6. Fluorination of ethylene with MnF3(T= 265→460ºС, contact time ≈7 min.).
2.4. Some features and possible mechanisms of C1-C2 hydrocarbons and C1-C2 polyfluoroalkanes fluorination with HVMFs
Based on the results obtained, several conclusions and assumptions have been made that can improve understanding of the processes of interaction of aliphatic compounds with HVMFs.
2.4.1. Probable mechanism of formation of 1,1-difluoroethane
The possibility of rearrangement of carbocation (I) to form 1,1-difluoroethane was confirmed (Scheme 2).

Scheme 2. Carbocation rearrangements with obtaining of 1,1-difluoroethane during cobalt trifluoride fluorination of ethylene.
For the first time, the suggestion of such a rearrangement was made by J. Burdon in 1976 [12] based on D. Clark's data on the stability of carbocations I and II (see [2]). It was found that during the fluorination of ethylene with CoF3, 1,1-difluoroethane had been detected in the reaction products in significant concentrations already at 40 ºC [5]. On the other hand, 1,2-difluoroethane itself did not react with CoF3 at temperatures below 50 ºC, meaning that isomerization of 1,2-difluoroethane to 1,1-difluoroethane at 40 ºC was excluded. Therefore, the conclusion that 1,1-difluoroethane forms via the carbocation rearrangement I→II seems logical.
It should be noted that this rearrangement I→II is typical for all HVMFs studied. Moreover, 1,2-difluoroethane in the reaction products with manganese trifluoride and cerium tetrafluoride was obtained in relatively low concentrations. This is likely due to the fact that the fluorination with manganese trifluoride and cerium tetrafluoride occurred at significantly higher temperatures, which could have increased the possibility of I→II rearrangement.
The results of fluorination of fluoroethane [13] may also be a certain confirmation of the possibility of the I→II rearrangement. Carbocation I can be formed during the fluorination of not only ethylene but also fluoroethane (CH2F-CH3). In monofluoroethane molecule, the electron density at the carbon atom bound to fluorine should be shifted toward the fluorine atom, therefore, the preferential electron abstraction followed by hydrogen substitution should occur at the carbon not bound to fluorine (Scheme 3).

Scheme 3.
However, the results of fluorination of fluoroethane with CoF3 at temperatures of 50-100 ºC and a contact time of 8 hours (S. Kurosawa et al. [13]) showed, as in reactions with ethylene, a predominant content of 1,1-difluoroethane in the reaction product (ratio CHF2-CH3: CHF2-CH2F ≈2÷4:1).
2.4.2. The effect of geminal fluorine atoms on hydrogen substitution
The results obtained confirmed that the ease of hydrogen substitution was reduced in the presence of geminal fluorine atoms (See [2]). As an example, the results of fluorination of 1,1-difluoroethane with cobalt trifluoride and cerium tetrafluoride were given (Table 1), which showed the predominant obtaining of 1,1,2-trifluoroethane (the ratio of 1,1,2-trifluoroethane to 1,1,1-trifluoroethane was ≈10÷20:1 for CoF3 depending on the reaction temperature). For CeF4, the ratio of trifluoroethane isomers was even more significant (the ratio of CHF2-CH2F to CF3-CH3 was ≈100:1).
Table 1. Results of fluorination of 1,1-difluoroethane with cobalt trifluoride and cerium tetrafluoride (contact time ~7 minutes) [4].
|
T, ºC |
HVMF |
Product yield (% vol.) |
|||||
|
CHF2-CH3 |
CHF2-CH2F |
CF3-CH3 |
CHF2-CHF2 |
CF3-CH2F |
CF3CHF2 |
||
|
97 |
CoF3 |
14,4 |
73,3 |
7,3 |
1,5 |
3,5 |
0,0 |
|
118 |
CoF3 |
0,1 |
76,0 |
3,1 |
7,5 |
12,1 |
1,2 |
|
290 |
CeF4 |
2,0 |
68,6 |
0,6 |
27,6 |
- |
0,2 |
The results obtained were explained by the fact that the fluorination of the 1,1-difluoroethane molecule with cobalt trifluoride (or other HVMFs) began with the abstraction of an electron by the Co+3 ion from the organic substrate. In 1,1-difluoroethane, the electron density at the fluorine-bonded carbon atom is strongly shifted toward the fluorine atoms, so a preferential electron abstraction followed by the elimination of an H+ from carbon not bound to fluorine should occur (Scheme 4) [6].

Scheme 4.
At the second step, radical IV could either be oxidized by a metal ion (Co3+) to form carbocation IV', or the radical was "quenched" through the transition of a fluorine atom from CoF3 [6]. A similar scheme applies to subsequent steps of fluorination.
2.4.3. Influence of fluorination conditions and the nature of HVMFs on the ratio of polyfluoroethanes isomers
The thesis that the substitution of hydrogen for fluorine in the intermediate steps of fluorination occurs predominantly at the carbon atom with a minimum number of attached fluorine atoms has been well confirmed by the results of fluorination on weak fluorinating agents: MnF3 and CeF4 (see Figs. 5 and 6). The results of fluorination with KCoF4 from [12] also do not contradict this conclusion. At the same time, during fluorination with CoF3, a high content of polyfluoroethanes with an asymmetric distribution of fluorine was observed.
It is known from the literature that carbocations (or similar radicals) with a positive charge at the carbon atom with the maximum number of attached fluorine atoms are more energetically favorable than their isomers with a positive charge at the carbon atom with a smaller number of attached fluorine atoms. As an example, we can cite the rearrangement +CH2CH2F→CH3CHF+ already discussed above (see 2.1.1) (Scheme 2), as well as the results of S. Kurosawa and colleagues [13-16]. In the article [14] calculations of the electronic structure of radicals are presented, which showed that the radical CH3CF2• (V, Scheme 4) is more energetically preferable than the radical CH2FCH2• (IV, Scheme 4). Similar calculations showed the energetic preference of [CFH2-CF2•] over [CF2H-CFH•][15].
In support of the calculated data, the article [13] presented the results of fluorination of ethane, fluoroethane, 1,1-difluoroethane and other polyfluoroethanes with cobalt trifluoride in the temperature range of 50-200 °C. The fluorination was carried out in a periodic mode, the reaction time was 8 hours (400 hours in one synthesis). The results of fluorination of 1,1-difluoroethane with cobalt trifluoride are shown in Table 2.
Table 2. Results of fluorination of 1,1-difluoroethane with cobalt trifluoride at a contact time of 8 hours (* contact time 400 hours) [13].
|
Reactant |
T, °C |
Product yield (%) |
|||||
|
CH3CF2H |
CFH2-CFH2 |
CF3CH3 |
CF2H-CFH2 |
CF3CFH2 |
CF2HCFH2 |
||
|
CH3CF2H |
50 |
86 |
0 |
11 |
2 |
1 |
0 |
|
CH3CF2H |
50 * |
4 |
0 |
58 |
12 |
25 |
1 |
|
CH3CF2H |
75 |
53 |
0 |
29 |
14 |
4 |
0 |
|
CH3CF2H |
100 |
6 |
0 |
34 |
49 |
7 |
4 |
When comparing the results in Tables 1 and 2, it can be noted that even at lower temperatures but with a significantly higher value of the contact time, the fluorination shifted towards the obtaining of 1,1,1-trifluoroethane, and an increase in the reaction time to 400 hours led to the obtaining of 1,1,1-trifluoroethane with an almost 60% yield and an isomer ratio of CF3CH3 :CF2H-CFH2≈5:1. The probable mechanism for the formation of 1,1,1-trifluoroethane through rearrangement with the migration of H+ (or the H• radical) is shown in Scheme 4.
Based on an analysis of the fluorination results of 1,1-difluoroethane, it can be assumed that fluorination with CoF3 created conditions that facilitated the rearrangement of carbocations (or radicals) to form more energetically favorable compounds. And one of the factors that could influence such rearrangements was the contact time of the organic compound with CoF3.
In this case, contact time should be understood as a general indicator of a set of processes, including: adsorption of an organic compound molecule on the surface of HVMFs, transport of fluorine from the inner lattice layers of HVMFs to the surface, chemical reaction, desorption of an organic compound molecule after fluorination, etc.
The hypothesis was experimentally tested by studying the dependence of the ratio of tetrafluoroethane isomers during the fluorination of ethylene with CoF3 at 250 ºC and different contact times (1÷7,5 sec.). The results are shown in Table 3 [4].
Table 3. Ratio of tetrafluoroethane isomers during the fluorination of ethylene (T=250ºC, contact times 1÷7,5 sec.) [4].
|
Time (sec.) |
Product yield (%) |
||||||||
|
CF2HCH3 (1) |
CFH2CFH2 (2) |
CF3CH3 (3) |
CF2HCFH2 (4) |
CF2HCF2H (5) |
CF3CFH2 (6) |
C2F5H (7) |
C2F6 (8) |
5/6 ratio |
|
|
bp -24,05 |
bp 26 |
bp -47,6 |
bp 5 |
bp -22,5 |
bp -26,5 |
bp -48,5 |
bp -78 |
||
|
1 |
0,5 |
1,0 |
0 |
85,0 |
9,0 |
4,0 |
0,5 |
0 |
2,25:1 |
|
2,5 |
0 |
0,5 |
1,0 |
56,0 |
24,0 |
15,0 |
3,0 |
0,5 |
1,6:1 |
|
7,5 |
0 |
0 |
1,0 |
47,0 |
26,3 |
19,5 |
6,5 |
0,7 |
1,34:1 |
These results also correlate quite well with the results of ethylene fluorination from the article [7] (Table 4).
Table 4. Results of fluorination and the ratio of tetrafluoroethane isomers at short contact times [7].
|
Т, ºC |
Time (sec.) |
Product yield (%) |
||||||||
|
CFH2CFH2 (2) |
CF2HCH3 (1) |
CF3CH3 (3) |
CF2HCFH2 (4) |
CF2HCF2H (5) |
CF3CFH2 (6) |
C2F5H (7) |
C2F6 (8) |
5/6 ratio |
||
|
150 |
1,4 |
19,9 |
8,1 |
0,7 |
70,3 |
1,8 |
0,0 |
0,0 |
0,0 |
|
|
2,8 |
1,0 |
0,3 |
0,9 |
86,9 |
5,6 |
5,0 |
0,3 |
0,0 |
1,12:1 |
|
|
350 |
1,0 |
0,0 |
0,0 |
0,2 |
5,2 |
20,0 |
10,7 |
43,2 |
20,7 |
1,86:1 |
|
2,0 |
0,0 |
0,0 |
0,0 |
1,4 |
10,6 |
6,0 |
40,0 |
42,0 |
1,76:1 |
|
Other possible pathways for the obtaining of 1,1,1,2-tetrafluoroethane, namely the reaction of HF elimination from 1,1,2-trifluoroethane with subsequent addition of fluorine to the double bond and isomerization of 1,1,2,2-tetrafluoroethane were not taken into account for the following reasons:
- Firstly, the elimination of HF is usually stimulated by increasing the reaction temperature, and in the reactions studied, the maximum content of 1,1,1,2-tetrafluoroethane should have been observed in reactions with CeF4, which did not correspond to the experimental results. In addition, 1,1,2-trifluoroethane is a fairly stable compound up to 300°C; therefore, under the process conditions, the elimination of hydrogen fluoride from 1,1,2-trifluoroethane is unlikely [6].
- Secondly, it was experimentally established that in the process of fluorination of 1,1,2,2-tetrafluoroethane with CoF3, only 1,1,2,2-tetrafluoroethane itself, pentafluoroethane and hexafluoroethane were observed in the reaction products. That is, isomerization did not occur [6].
The results obtained showed (Tables 3 and 4) that with increasing contact time, the ratio between the isomers shifted toward 1,1,1,2-tetrafluoroethane, which may serve as some confirmation of the hypothesis. Increasing the contact time to approximately 300-360 sec. (6-7 min.) shifted the ratio of CF2HCF2H к CF3CFH2 to ≈0.6:1.
The mechanism that explained the appearance of 1,1,1,2-tetrafluoroethane was proposed in the article [6] and consisted of the possibility of rearrangements of the intermediate radical VI (or carbocation VII), which led to the appearance of an asymmetric isomer (Scheme 5). The same rearrangement of radical VI (or carbocation VII) can explain also the appearance of 1,1,1,2-tetrafluoroethane in the products of fluorination of acetylene with cobalt trifluoride (see 2.2).

Scheme 5. Carbocation (radical) rearrangements with obtaining of 1,1,1,2-tetrafluoroethane [6].
Another parameter affecting the isomer ratio was the nature of the fluorinating reagent. It was found that when fluorination was carried out on the same equipment under comparable conditions (for example, at the maximum yield of tetrafluoroethanes), the ratio of the concentrations of the asymmetric isomer (CF3CFH2) to the symmetric isomer (CF2HCF2H) was 1,5÷1,7:1 for strong cobalt trifluoride, 0,25÷0,4:1 for weaker manganese trifluoride, and 0,01÷0,02:1 for even weaker cerium tetrafluoride [3,4]. These results are in good agreement with the dependence of the strength of the fluorinating reagent on the oxidation potential of the metal ion (see the 1st part of the communication [1]).
CoF3> MnF3> CeF4
To summarize the above, it can be assumed with a high degree of probability that the use of strong HVMFs and an increase in the contact time will facilitate the rearrangement of intermediate radicals (carbocations) with the formation of asymmetric polyfluoroalkanes. In the articles of J. Burdon, this process is also called H-migration (see [2]). Conversely, a decrease in the contact time in the case of strong HVMFs and the use of weak HVMFs will lead to the preferential formation of polyfluoroalkanes with a more symmetrical distribution of fluorine atoms.
An additional factor affecting the distribution of isomers may be the different adsorption tendency of symmetric and asymmetric fluoroethanes on the surface of cobalt trifluoride particles due to the significant difference in boiling points (BP values see Table 3). It was suggested in [7] that difluoroethanes and trifluoroethanes with a more symmetrical distribution of fluorine atoms have a greater tendency to such adsorption.
Thus, according to the authors, the rate of conversion of low-boiling and weakly adsorbing 1,1-difluoroethane (BP ≈-24°C) will be determined by the rate of desorption of high-boiling products from the surface of cobalt trifluoride. In turn, for 1,2-difluoroethane with a relatively high boiling point (≈+26°C) and higher adsorption capacity, the rate of conversion will be determined by the rate of chemical reaction with CoF3. Accordingly, the rate of further conversion of 1,2-difluoroethane will always be higher than the rate of conversion of 1,1-difluoroethane, therefore, with an increase in the contact time, the ratio of difluoroethane isomers will shift towards the predominance of 1,1-difluoroethane due to its slower conversion to trifluoroethanes.
As a commentary on this hypothesis, it can be noted that the ability to adsorb can affect the composition of the fluorination products, but only in a limited temperature range and at certain steps of the process, since:
- As the fluorination temperature increases, the influence of adsorption capacity factors should decrease.
- Adsorption capacity cannot affect, for example, the ratio of tetrafluoroethane isomers in any way, since fluorination, except for very long contact times, occurs almost exclusively via the relatively high-boiling 1,1,2-trifluoroethane (BP = +5°C).
2.4.4. Mechanism of oligomerization during fluorination with cerium tetrafluoride
A characteristic feature of the fluorination of ethylene with cerium tetrafluoride was the formation of significant quantities of compounds that are liquid under normal conditions (up to 30% by weight of the collected products) in the reaction products at temperatures from 150 to 270 °C. The analysis of these liquid products showed the presence of polyfluoroalkenes and polyfluoroalkanes with a number of carbon atoms of 4 or more.
In the article [6], a hypothesis was put forward that at temperatures of 150÷270°C, the formation of oligomeric compounds occurs by the mechanism of ionic polymerization, when the formed carbocation I could have a sufficient lifetime to enter into a competing process with ethylene to obtain oligomers (Scheme 6). At higher temperatures, the reaction of "quenching" with fluorine (with possible rearrangements similar to Scheme 2) became the main pathway.

Scheme 6. Possible mechanism of oligomerization during ethylene fluorination with cerium tetrafluoride [6].
This mechanism of oligomerization can also be explained from the point of view of the cation-radical theory of fluorination, considered in the first part of the communication [1]. The direction of fluorination processes is determined by the time required to bring a fluorine species from HVMF lattice to the surface. And this time is different for the different fluorides. For example, in the case of fluorination of arenes with CsTlF4 (or CsCoF4), additional spatial difficulties in delivering fluorine to the surface created the opportunities for 1,2-migration of fluorine with obtaining of fluorinated aromatic compounds. Fluorination of benzene over CeF4 at 480°C also resulted in the obtaining of a significant amount of polyfluorobenzenes with 3–4 fluorine atoms [17], which may also indicate the presence of spatial difficulties in the transportation of fluorine to the surface in CeF4 lattice. Accordingly, in the process of ethylene fluorination with CeF4, the lifetime of cation I (or the corresponding radical) can be long enough to allow a side polymerization reaction, yielding oligomeric products. A similar mechanism for obtaining of oligomers can be applied to the fluorination of acetylene with CeF4 (see 2.2).
2.4.5. Possible mechanism for obtaining of 1,1,2-trifluoroethane at low temperatures during fluorination with cobalt trifluoride
When studying the fluorination of ethylene with cobalt trifluoride at low temperatures (20-60 °C), attention was paid to the presence of large quantities of 1,1,2-trifluoroethane in the reaction mixture (20-30%) [5]. At the same time, it was experimentally found that 1,2-difluoroethane and 1,1-difluoroethane did not react with cobalt trifluoride at such low temperatures.
To explain the obtained results, it was suggested that at temperature range 20÷60 °C fluorination may partially occur similarly to the fluorination of aromatic compounds.
For example, when benzene was fluorinated with cobalt trifluoride, the process proceeded through the obtaining of fluorobenzene, and then 1,4-difluorobenzene. Only at the next step was the addition of fluorine to the aromatic ring bonds observed (considered in detail in [1]). If we assume that in the case of fluorination of ethylene at low temperatures, at the initial step the process could partially proceed through the substitution of hydrogen with the formation of monofluoroethylene followed by the addition of fluorine at the double bond (Scheme 7), then the appearance of large quantities (20-30%) of 1,1,2-trifluoroethane becomes possible.

Scheme 7.
The hypothesis is indirectly supported by the fact that monofluoroethylene was detected in traces in the reaction products of ethylene with another HVMF, namely, cerium tetrafluoride. The possibility of a substitution reaction was also suggested in [4] when considering the fluorination of acetylene with CeF4, with the possible formation of monofluoroacetylene in the first step of fluorination. However, it should be noted that there is no clear experimental confirmation of the formation of CHF=CH2 during fluorination with cobalt trifluoride.
2.4.6. Possible influence of hydrogen fluoride and the CoF3→CoF2 transition on fluorination processes
The first suggestion about the possible catalytic influence of hydrogen fluoride on the fluorination processes with cobalt trifluoride was made in [3]. The influence of HF on the fluorination of difluoromethane, trifluoromethane and pentafluoroethane was studied in more detail in the article [8].
Based on the experimental results, the authors of [8] suggested that at temperatures of 185÷205 °C, the reaction mechanism could change and up to 300°C fluorination could proceed via two competing mechanisms. The cause of such changes could be the transition through the critical temperature of HF (188ºC) and, as a result, a change in the aggregate state of hydrogen fluoride even with a slight change in temperature.
These suggestions were based on the fact that HF has a high adsorption capacity (under normal conditions, one mole of cobalt fluorides can hold on the surface up to 0,3 moles of HF). Therefore, during the fluorination process, a thin film of HF could form on the surface of cobalt trifluoride. In this case, the change in the state of aggregation of HF upon passing through the critical temperature could lead to the desorption of most of the HF from the surface. After this, the surface of the cobalt fluorides particles was cleaned, and the gaseous reactants interacted directly with the fluorinating reagent. The authors explained the observed decrease in the reaction rate upon passing the critical temperature of HF by the fact that, since HF is a catalyst, decreasing its concentration on the reaction surface also reduced the reaction rate. Above 300 ºC, hydrogen fluoride completely desorbed, and its effect on the process ceased.
A catalytic mechanism was also proposed, which involved adsorbed hydrogen fluoride forming hydrogen bonds with adsorbed molecules of gaseous reactants, thereby reducing the electron-withdrawing activity of fluorine atoms (Scheme 8). This, in turn, facilitated the removal of an electron from a carbon atom, followed by the formation of a cation radical.

Scheme 8.
In conclusion, it was proposed that hydrogen fluoride could participate in two processes simultaneously during fluorination. On the one hand, HF adsorbed on the surface of cobalt trifluoride could have acted as a catalyst; on the other hand, the accumulation of HF on the surface led to diffusional difficulties in transporting gaseous reactants to the reaction surface and to blocking of the reaction centers.
Furthermore, experimental results showed that the fluorination process is not slowed down by the diffusion of gaseous reagents through the layer of forming CoF2. The absence of diffusion limitations was explained by the fact that, since the density of cobalt difluoride is higher than that of cobalt trifluoride, cracks constantly form in the solid layer, preventing diffusion limitations [8].
2.5. Fluorination of organic compounds using complex nickel and copper fluorides
Fluorination with complex fluorides of nickel (Ni+4) and copper (Cu+3) in reactions with hydrocarbons was considered in [9]. It should be noted that, unlike most HVMFs, these complex fluorides can be prepared from commonly available starting materials (nickel and copper chlorides). K2NiF6 and K3CuF6 were prepared directly in the reactor from mixtures of potassium fluoride with CuCl or NiCl2, respectively, by treatment with elemental fluorine.
The fluorinating properties of K2NiF6 and K3CuF6 were studied in processes with 1,1,1,2-tetrafluoroethane, hexafluoropropylene, and several hydrocarbons. The authors concluded that complex fluorides could be used for the fluorination of alkanes and alkenes.
References
- Kornilov V. V., Fluorination with high valency metal fluorides, cation-radical theory of fluorination of aromatic compounds, Fluorine notes, Iss. 5(162), http://en.notes.fluorine1.ru/public/2025/5_2025/article_1.html, http://dx.doi.org/10.17677/fn20714807.2025.05.01.
- Kornilov V. V., Possible mechanisms of fluorination of aliphatic hydrocarbons with high valency metal fluorides, Fluorine notes, Iss. 6(163), http://en.notes.fluorine1.ru/public/2025/6_2025/article_1.html, http://dx.doi.org/10.17677/fn20714807.2025.06.0.1.
- Asovich V.S., Kornilov V.V., Kostajev R.A., Maximov B.N., Fluorination of methane and its fluoroderivarives by cobalt trifluoride, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 1, 1994, p. 103-107.
- Asovich V.S., Kornilov V.V., Kostajev R.A., Mel’nichenko B.A., Maruev A.V., Maximov B.N., Report “Fluorination of hydrocarbons by highest fluorides of cobalt, manganese and cerium”, The International Conference “Chemistry, Technology and Application of Fluorocompounds” (CTAF’94), Saint Petersburg, Russia, May 30- June 03, 1994.
- Asovich V.S., Kornilov V.V., Kostajev R.A., Mel’nichenko B.A., Maruev A.V., Maximov B.N., Fluorination of hydrocarbons by highest fluorides of cobalt, manganese and cerium, Zhurnal Organichnoi Khimii (Russian Journal of Organic Chemistry), Vol. 30, Is. 8, 1994, p. 1221.
- Asovich V.S., Kornilov V.V., Maximov B.N., Fluorination of ethylene by highest fluorides of cobalt, manganese and cerium, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 1, 1994, p. 107-111.
- Kostajev R.A., Pashkevich D.A., Desorption of reaction products during the process of Ethylene Fluorination with Cobalt Trifluoride, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 10, 1994, pp. 1624-1628; Костяев Р.А, Пашкевич Д.С., Десорбция продуктов реакций в процессе фторирования этилена трифторидом кобальта, ЖПХ, 1994, Т. 67, Вып. 10, с. 1624-1628.
- Kostajev R.A., Pashkevich D.A., The role of hydrogen fluoride and cobalt difluoride in the processes of hydrocarbon fluorination with cobalt trifluoride, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 12, 1994, pp. 2012-2016; Костяев Р.А, Пашкевич Д.С., Роль фтороводорода и дифторида кобальта в процессах фторирования углеводородов трифторидом кобальта, ЖПХ, 1994, Т. 67, Вып. 12, с. 2012-2016.
- Maximov B.N., Kornilov V.V., Kostajev R.A., Kosareva L.N., Complex Fluorides of Nickel- and Copper- as a Reagents for Fluorination of Hydrocarbons, The 2nd International Conference “Chemistry, Technology and Application of Fluorocompounds” (CTAF’97), Saint Petersburg, Russia, September 23-26, 1997.
- Kornilov V.V., Kostajev R.A., Maximov B.N., Mel’nichenko B.A., Fiodorova T.E, Fluorination organic compounds by Cobalt Trifluoride. Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 68, Iss 9, 1995, p. 1409-1417.
- Burdon J., Garnier L, Powell R. L., Fluorination of propane and propene over cobalt (III) trifluoride and potassium tetrafluorocobaltate (III), J. Chem. Soc., Perkin Trans. 2, 1996, Iss. 4., pp. 625-631, http://dx.doi.org/10.1039/P29960000625.
- Burdon J.,Knights J., Parsons I, Tatlow J. The fluorination of ethane and ethene over potassium tetrafluoride cobaltate (III) and cobalt trifluoride, Tetrahedron, 1976, V.32, p.1041-1043, https://doi.org/10.1016/S0040-4020(01)83232-4.
- Kurosawa S., Sekiya A., Arimura T., Suga A., The monofluorination of hydrofluorocarbons over cobalt trifluoride, Journal of Fluorine Chemistry, Volume 62, Issue 1, 1993, Pages 69-76, ISSN 0022-1139, https://doi.org/10.1016/S0022-1139(00)80082-2.
- Kurosawa S., Sekiya A., Arimura T., Suga A., Ab Initio Molecular Orbital Study on Monofluorination Reaction of Hydrofluorocarbons Over Cobalt Trifluoride, Journal of Japan Oil Chemists' Society, 1994, p. 650-652, DOI:10.5650/jos1956.43.650.
- Kurosawa S., Amimura T. Theoretical study of monofluorination reaction, Chem. Express., 1992, Vol.7, Iss. 6, p.429.
- Kurosawa S., Synthesis and Application of Fluorine-Containing Compounds, Journal of Japan Oil Chemists' Society, 1999, Vol. 48, Iss. 11, p. 1247, DOI: 10.5650/jos1996.48.1247
- Hudson A.G., Pedler A.E., Tatlow J.C., The fluorination of benzene over cerium tetrafluoride, Tetrahedron, 1969, Vol. 25, Iss. 18, pp. 4371-4374, https://doi.org/10.1016/S0040-4020(01)82976-8.
ARTICLE INFO
Received 19 December 2025
Accepted 12 February 2026
Available online February
2026
Recommended for publication by PhD M.A. Manaenkova
eLIBRARY Document Number (EDN) UOBVFM

Fluorine Notes, 2026, 164, 1-2
