Received: June 2023
DOI 10.17677/fn20714807.2023.04.01
Fluorine Notes, 2023, 149, 1-2
4,4,4-TRIFLUORO-2-BUTENONITRILE IN C-ALKYLATION REACTIONS OF INDOLES AND PYRROLES
A. L. Sigan, A. Yu. Volkonskii, N. D. Kagramanov, E. V. Guseva, N. D. Chkanikov
A. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Build. 1, 28 ul. Vavilova, 119334 Moscow, Russian Federation.
Fax: +7-(499) 135-5085; E-mail: asigan@yandex.ru
Annotation. Cyanoalkylation of indoles and pyrroles using 4,4,4-trifluoro-2-butenonitrile in the presence of aluminium bromide was used to synthesize 4,4,4-trifluoro-substituted 3-(indol-3-yl)- and 3-(pyrrol-2-yl)butanonitrites, which are converted by saponification of the nitrile group into 3 heteroaryl-substituted 4,4,4-trifluorobutyric acids.
Keywords. 4,4,4-trifluoro-2-butenonitrile, cyanoalkylation, Friedel-Crafts reaction, Lewis acids, nitriles, indoles, pyrroles, saponification.
Introduction
The results of our long-term systematic research in the field of new fluorine-containing bioactive compounds preparation show that various CF3-substituted heterocycles can be synthesized using highly electrophilic 2- and/or 3-substituted derivatives of 4,4,4-trifluoro-2-butenonitrile. The key step in these transformations is the addition of nucleophiles to the double bond of fluorine-containing alkenes. This reaction proceeds exclusively at the β-position relative to the CN group. In this case, transformations with the formation of a new C–C bond are of greatest interest from the standpoint of organic chemistry. The 2-cyano-, 2-alkoxycarbonyl- and 2-diethoxyphosphoryl- derivatives of 4,4,4-trifluoro-2-butenonitrile we have studied earlier are capable to C-alkylate similarly a number of π-donor aromatic and heteroaromatic compounds at room temperature in the absence of a catalyst [1]. In this work, we studied the reactivity of the simplest representative of the considered class of electrophiles, 4,4,4-trifluoro-2-butenonitrile, CF3CH=CHCN (1).
The alkene 1 is synthetically available and structurally attractive as a building block in the synthesis of new fluorine-containing compounds. However, until now its use has been limited to individual reactions of Michael addition and cycloaddition [2-6]. The study of the reactions’ conditions of heterocycles C-alkylation should significantly expand the possibilities of 4,4,4 trifluoro-2-butenonitrile 1 practical using. Indoles and pyrroles were chosen as model heterocycles because they easily react with similar transformations, and the products of these transformations can serve as starting materials for obtaining new bioactive substances [7].
Results and discussion
As expected, the reactivity of butenonitrile 1 with respect to π-donor heterocycles was significantly lower than that of previously studied fluorine-containing 1,1-dicyanoethylenes, which C-alkylated indole and pyrrole derivatives under mild conditions [8–11]. It was found that alkene 1 doesn’t interact with N-methylindole and N-methylpyrrole in various solvents (benzene, dioxane, chloroform, CCl4) even when heated in sealed ampoules in an inert atmosphere to 120°C. At higher temperatures only decomposition products of starting materials were found.
Due to the low reactivity of the starting butenonitrile 1, we decided to use Friedel-Crafts C‑alkylation in presence of Lewis acids (see, for example, [12]), which is widely used in electrophilic substitution reactions involving heterocycles. This method in most cases has a number of limitations, in particular, low yields and selectivity, high requirements for anhydrous conditions and long reaction times have been noted. Also in our case, during the interaction of butenonitrile 1 and N-methylindole or N-methylpyrrole in the presence of AlBr3 in СH2Br2 in an inert atmosphere under standard conditions of the Friedel-Crafts reaction (i. e. with rapid sequential mixing of all reagents), the target products of cyanoalkylation were obtained in low yields (up to 25%). Characteristic for indoles and pyrroles transformation in the presence of Lewis acids, the products of resinization and dimerization of heterocycles were also found [13–15].
At the same time, it was found that preliminary mixing of butenonitrile 1 and Lewis acid, stirring until homogenization, subsequent addition of heterocycles and then keeping the reaction mixture until complete conversion of the starting materials can significantly reduce the by-products formation. In this way it became possible to obtain 4,4,4-trifluoro-3-(indol-3-yl)- and -3-(pyrrol-2-yl)butanonitriles 2a-c and 3a-c in up to 73% yields (Scheme 1).
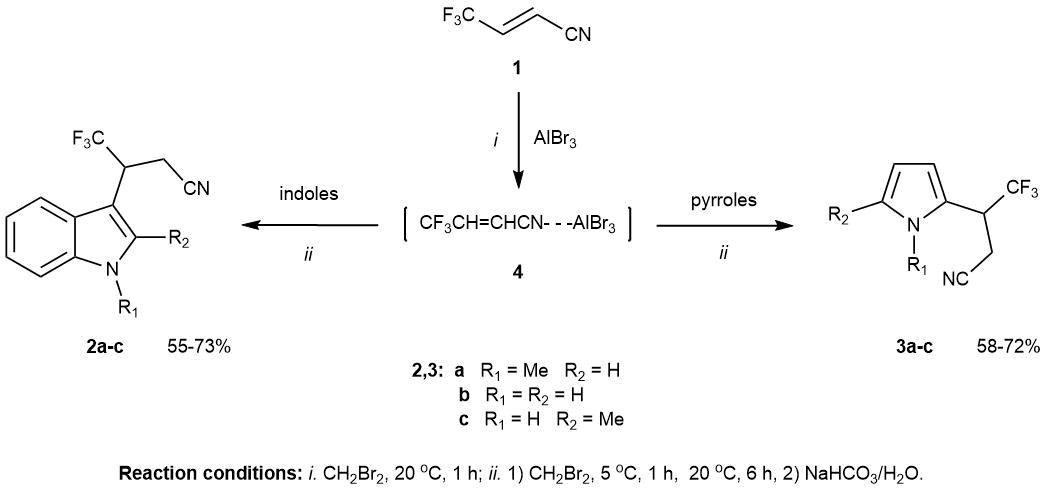
Scheme 1. Supposed route for cyanoalkylation of indoles and pyrroles by butenonitrile 1 in the presence of AlBr3.
Apparently, in this case, butenonitrile 1 forms a stable complex [CF3CH=CHCN→AlBr3] 4 with aluminium bromide (Scheme 1), which activates the β-position of double bond and deactivates the Lewis acid, reducing content of its free form in solution.
It should be noted that in the general case, the chemical properties of nitrile-Lewis acid complexes are characterized by a greater ease of nucleophilic attack on the nitrile carbon atom compared to free nitriles. The reactivity of α,β-unsaturated nitriles containing additional electrophilic substituents at its double bond has been less studied. The direction of the nucleophilic attack in such compounds should depend on the electrophilic substituent on the double bond [16]. In our case, the reaction center in the starting butenonitrile 1 has been the carbon atom adjacent to the trifluoromethyl group. This is evidenced by the 19F NMR spectra of the obtained products 2 and 3, where the trifluoromethyl group appears as a doublet rather than a triplet, as might be expected in the reverse addition. The products structure was also confirmed by mass spectrometry data.
When trying to identify and characterize the active intermediate 4, only indirect evidence of its formation was obtained. Thus, after mixing butenonitrile 1 with AlBr3 in СH2Br2 in an equimolar ratio under standard reaction conditions (20°C, 1 hour) and subsequent removal of the solvent in a water jet pump vacuum, a hygroscopic powder was obtained. The mass of the residue was practically corresponded to the total mass of the used reagents. Analysis of IR spectra showed a shift of the C≡N group stretching bands to higher frequencies (2307 cm-1) in comparison with the spectrum of the initial nitrile (2240 cm-1) [17]. This is considered in the literature as reliable criterion for the participation of nitrile nitrogen in the formation of a coordination bond [18]. After hydrolysis of resulting powder, extraction with CH2Br2, and analysis of the organic phase by gas chromatography, the starting butenonitrile 1 was identified therein. The reaction of the resulting powder with heterocycles under standard conditions led to the formation of the same cyanoalkylated compounds.
Heterocyclic compounds containing easily modified functional groups are of great interest for fine organic synthesis and drug development. In our work, to expand the bank of functionally substituted fluorine-contaning indoles and pyrroles, we used the well-known transformation of the nitrile group - the saponification reaction with the formation of the corresponding carboxylic acids 5a-c and 6a-c (Scheme 2).
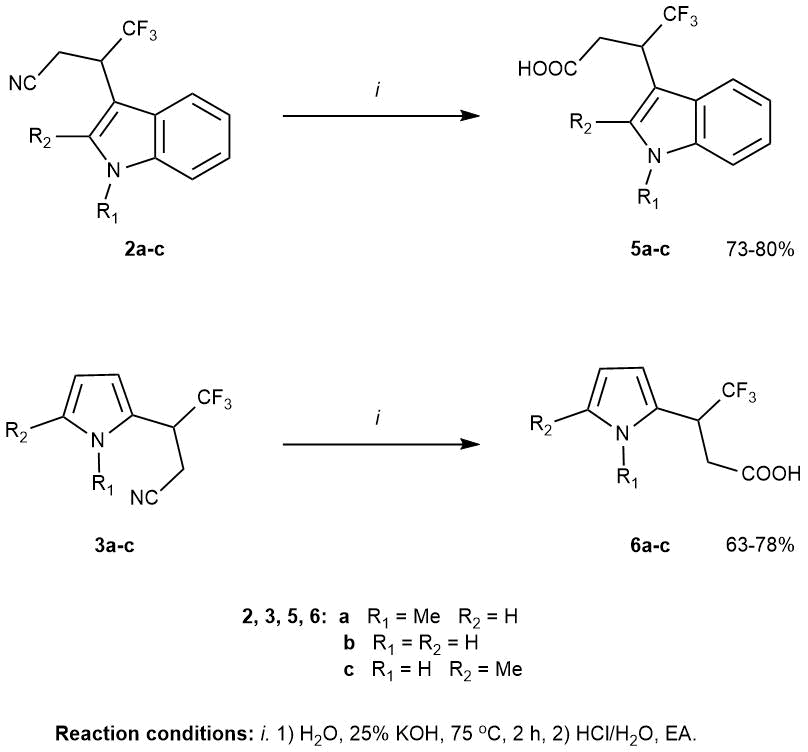
Scheme 2. Saponification of the nitrile group in the products of cyanoalkylation of indoles and pyrroles.
It should be noted that, from our point of view, all products obtained using butenonitrile 1 as a building block are of independent interest as potentially biologically active compounds and are promising for building more complex condensed systems (cf. Ref. [19-21]).
Experimental section
1H and 19F NMR spectra were recorded on a Bruker Avance TM400 spectrometer with operating frequencies of 400.13 and 376.50 MHz, respectively. The chemical shifts of the 1Н nuclei were determined relative to the residual proton signal of the solvent for solutions in CDCl3 (δН 7.26 ppm) and in DMSO-d6 (δН 2.50 ppm), 19F nuclei - relative to CFCl3 as an external standard.
The mass spectra of the compounds were recorded on a Finnigan Polaris Q mass spectrometer (ion trap, EI, 70 eV). Compound samples in solid form or in the form of a 1% solution in CH2Cl2 (0.2 μl) were loaded into quartz microampoules, which were inserted into the heated tip of the direct injection rod. Thermal mass spectrograms were taken in the range from 30 to 200°С.
IR spectra were recorded on a Bruker spectrometer (Tensor 37) in the region of 700–3600 cm‑1 (in tablets with KBr).
Thin-layer chromatography (TLC) was performed on TLC Silica gel 60 F254 plates (Merck) using petroleum ether (PE) - ethyl acetate (EA) = 3:1 for the nitriles 2a-c and 3a-c or PE-EA-MeOH = 20:10:1 as eluents for the acids 5a-c and 6a-c. The products were purified by column chromatography using silica gel Merck Kieselgel 60.
Solvents, if necessary, were absolutized according to standard methods. In the syntheses, Aldrich reagents were used. 4,4,4-trifluoro-2-butenonitrile 1 was obtained according to the procedure [4].
Reaction of trifluorobutenonitrile 1 with indole and pyrrole derivatives.
Synthesis of 4,4,4-trifluoro-3-(N-methylindol-3-yl)butanonitrile (2a).
Standard procedure.
30 ml of CH2Br2, 3.0 g of AlBr3 (11.2 mmol) and 2.03 g (16.8 mmol) of butenonitrile 1 were successively placed in a two-necked 50 ml flask under argon atmosphere. The resulting solution was stirred for 1 h until a homogeneous state, cooled to 5°С and then the solution of 2.2 g (16.8 mmol) of N-methylindole in 2.0 ml of CH2Br2 was added dropwise. The resulting mixture was stirred for 1 h, warmed to room temperature, and the resulting homogeneous solution was stirred for 6 h until complete conversion of the starting N-methylindole (TLC control). Reaction mixture was hydrolyzed with saturated NaHCO3 solution to pH = 8 and extracted with CH2Br2 (2 x 30 ml). The organic layer was dried over Na2SO4, the solvent was removed in vacuum, the residue was chromatographed on a silica gel column (eluent PE-EA = 3:1) and 3.1g of a colorless powder was obtained (yield 73%), Rf = 0.35.
1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 3.06 (2H, d, 7.70 Hz, CH2); 3.81 (3H, s, CH3); 4.00-4.20 (1H, m, CHCF3); 7.23 (1H, s, Ar); 7.23 (1H, t, 6.99 Hz, Ar); 7.30 (1H, t, 7.31 Hz, Ar); 7.41 (1H, d, 8.28 Hz, Ar); 7.63 (1H, d, 7.98 Hz, Ar).
19F NMR spectrum (CDCl3, δ, ppm, J/Hz): -70.51 (3F, d, 8.11 Hz, CF3).
Mass spectrum (m/z (Irel (%)): 252 [M]+ (28), 212 [M-CH2CN]+ (100), 192 [M-CH2CN-HF]+ (6), 183 [M-CF3]+ (4), 162 [M-CH2CN-CF2]+ (21).
4,4,4-Trifluoro-3-(indol-3-yl)butanonitrile (2b).
Colorless powder, 55% yield, Rf = 0.30. (colorless oil, see Ref. [19]).
1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 3.07 (2H, d, 6.99 Hz, CH2); 4.00-4.16 (1H, m, CHCF3); 7.24 (1H, t, 6.99 Hz, Ar); 7.26 (s, 1H, Ar); 7.30 (1H, t, 7.31 Hz, Ar); 7.41 (1H, d, 7.95 Hz, Ar); 7.62 (1H, d, 7.63 Hz, Ar); 8.45 (1H, s, NH) (1Н NMR spectrum in acetone-d6 see Ref. [19]).
19F NMR spectrum (CDCl3, δ, ppm, J/Hz): -70.51 (3F, br.s, CF3).
Mass spectrum (m/z (Irel (%)): 238 [M]+ (46), 198 [M-CH2CN] (100), 178 [M-CH2CN-HF]+ (52), 169 [M-CF3]+ (8) (mass spectrum, Ref. [19]).
4,4,4-Trifluoro-3-(2-methylindol-3-yl)butanonitrile (2c).
Colorless powder, 62% yield, Rf = 0.35.
1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 2.37 (3H, s, CH3); 3.17 (1H, d.d, 5.75 Hz, 17.5 Hz, CH(а)Н(b)); 3.50 (1H, d.d, 9.90 Hz, 17.5 Hz, CH(а)Н(b)); H(a)/H(b) – AB system; 4.04-3.94 (1H, m, CHCF3); 7.19 (1H, t, 7.21 Hz, Ar); 7.23 (1H, t, 7.09 Hz, Ar); 7.31 (1H, d, 8.44 Hz, Ar); 7.56 (1H, d, 7.58 Hz, Ar); 8.20 (1H, s, NH).
19F NMR spectrum (CDCl3, δ, ppm, J/Hz): -69.62 (3F, d, 7.85 Hz, CF3).
Mass spectrum (m/z (Irel (%)): 252 [M]+ (50), 212 [M-CH2CN]+ (100), 192 [M-CH2CN-HF]+ (60), 162 [M-CH2CN-CF2]+ (20).
4,4,4-Trifluoro-3-(N-methylpyrrol-2-yl)butanonitrile (3a).
Colorless powder, 70% yield, Rf = 0.44.
1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 2.93 (1H, d.d, 9.14 Hz, 16.90 Hz, CH(a)H(b)); 3.03 (1H, d.d, 5.94 Hz, 16.90 Hz, CH(а)Н(b)); H(a)/H(b) – AB system; 3.67 (3H, s, CH3); 3.85-3.98 (1H, m, CHCF3); 6.25 (1H, m, Ar); 6.34 (1H, m, Ar); 6.73 (1H, m, Ar).
19F NMR spectrum (CDCl3, δ, ppm, J/Hz): -71.16 (3F, d, 8.25 Hz, CF3).
Mass spectrum (m/z (Irel (%)): 202 [M]+ (44), 162 [M-CH2CN]+ (100), 142 [M-CH2CN-HF]+ (8), 133 [M-CF3] + (4), 112 [M-CH2CN-CF2]+ (23).
4,4,4-Trifluoro-3-(pyrrol-2-yl)butanonitrile (3b).
Colorless powder, 58% yield, Rf = 0.38.
1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 2.90 (1H, d.d, 8.58 Hz, 17.01 Hz, CH(a)H(b)); 3.00 (1H, d.d, 5.40 Hz, 17.01 Hz, CH(a)H(b)); H(a)/H(b) – AB system; 3.77-3.86 (1H, m, CHCF3); 6.26 (1H, m, Ar); 6.30 (1H, m, Ar); 6.84 (1H, m, Ar); 8.51 (1H, s, NH).
19F NMR spectrum (CDCl3, δ, ppm, J/Hz): -70.77 (3F, d, 8.25 Hz, CF3).
Mass spectrum (m/z (Irel (%)): 188 [M]+ (45), 148 [M-CH2CN]+ (92), 128 [M-CH2CN-HF]+ (100), 119 [M-CF3]+ (9).
4,4,4-Trifluoro-3-(5-methylpyrrol-2-yl)butanonitrile (3c).
Colorless powder, 72% yield, Rf = 0.44.
1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 2.28 (3H, s, CH3); 2.90 (1H, d.d, 8.27 Hz, 16.66 Hz, CH(а)Н(b)); 2.97 (1H, d.d, 5.72 Hz, 16.66 Hz, CH(a)H(b)); H(a)/H(b) – AB system; 3.72-3.85 (1H, m, CHCF3); 5.91 (1H, m, Ar); 6.16 (1H, m, Ar); 8.16 (1H, s, NH).
19F NMR spectrum (CDCl3, δ, ppm, J/Hz): -70.84 (3F, d, 7.85 Hz, CF3).
Mass spectrum (m/z (Irel (%)): 202 [M]+ (45), 162 [M-CH2CN]+ (88), 142 [M-CH2CN-HF]+ (100), 133 [M-CF3]+ (9).
Saponification of the nitrile group.
Synthesis of 4,4,4-trifluoro-3-(N-methylindol-3-yl)butanoic acid (5a).
Standard procedure.
To 20.0 ml of a 25% aqueous solution of KOH was added 1.6 g (6.4 mmol) of 4,4,4-trifluoro-3-(N-methylindol-3-yl)butanonitrile (2a), the resulting suspension was heated to 75 °C, kept up to formation of a homogeneous solution and stirred for another 2 hours. The mixture was neutralized with HCl (conc.), extracted with EA (2 x 50 ml) and dried over Na2SO4. After column chromatography (eluent PE–EA–MeOH = 20:10:1), the organic solvent was removed, the residue was recrystallized from a mixture of PE–CH2Cl2 = 3:1 and 1.3 g of a colorless powder was obtained (yield 77%), Rf = 0.33.
1H NMR spectrum (DMSO-d6, δ, ppm, J/Hz): 3.27-3.40 (2H, m, CH2); 3.81 (3H, s, CH3); 4.50‑4.60 (1H, m, CHCF3); 7.11 (1H, t, 7.23 Hz, Ar); 7.22 (1H, t, 7.23 Hz, Ar); 7.48 (1H, d, 8.17 Hz, Ar); 7.56 (1H, s, Ar); 7.72 (1H, d, 7.93 Hz, Ar); 12.47 (1H, s, COOH) (1Н NMR spectrum in acetone-d6 see Ref. [20]).
19F NMR spectrum (DMSO-d6, δ, ppm, J/Hz): -69.61 (3F, br.s, CF3).
Mass spectrum (m/z (Irel (%)): 271 [M]+ (63), 251 [M-HF]+ (46), 223 [M-HF-CO]+ (15), 212 [M‑CH2COOH]+ (100), 162 [M-CH2COOH-CF2] (28) (mass spectrum cf. Ref. [20]).
4,4,4-Trifluoro-3-(indol-3-yl)butanoic acid (5b).
Colorless powder, 73% yield, Rf = 0.28.
1H NMR spectrum (DMSO-d6, δ, ppm, J/Hz): 2.97 (1H, d.d, 9.40 Hz, 16.03 Hz, CH(a)H(b)); 3.05 (1H, d.d, 5.20 Hz, 16.03 Hz, CH(а)Н(b)); H(a)/H(b) – AB system; 4.23-4.34 (1H, m, CHCF3); 7.05 (1H, t, 7.19 Hz, Ar); 7.13 (1H, t, 7.19 Hz, Ar); 7.40 (1H, d, 8.07 Hz , Ar); 7.47 (1H, s, Ar); 7.63 (1H, d, 7.96 Hz, Ar); 11.24 (1H, s, NH); 12.50 (1H, s, COOH) (1Н NMR spectrum in acetone-d6 see Ref. [19]).
19F NMR spectrum (DMSO-d6, δ, ppm, J/Hz): -69.67 (3F, br.s, CF3).
Mass spectrum (m/z (Irel (%)): 257 [M]+ (81), 237 [M-HF]+ (31), 198 [M-CH2COOH]+ (100), 188 (25) [M-CF3]+ (mass spectrum, cf. Ref. [19]).
4,4,4-Trifluoro-3-(2-methylindol-3-yl)butanoic acid (5c).
Colorless powder, 80% yield, Rf = 0.33.
1H NMR spectrum (DMSO-d6, δ, ppm, J/Hz): 2.40 (3H, s, CH3); 3.02-3.13 (2H, m, CH2); 4.09-4.26 (1H, m, CHCF3); 6.98 (1H, t, 7.23 Hz, Ar); 7.04 (1H, t, 7.00 Hz, Ar); 7.30 (1H, d, 7.70 Hz, Ar); 7.50 (1H, d, 7.70 Hz, Ar); 11.12 (1H, s, NH); 12.44 (1H, s, 1H, COOH) (1Н NMR spectrum in acetone-d6 see Ref. [21]).
19F NMR spectrum (DMSO-d6, δ, ppm, J/Hz): -68.52 (3F, br.s, CF3).
Mass spectrum (m/z (Irel (%)): 271 [M]+ (71), 251 [M-HF]+ (11), 212 [M-CH2COOH]+ (100), 202 [M-CF3]+ (25), 192 [M-HF-CH2COOH]+ (8), 162 [M-CH2COOH-CF2]+ (19) (mass spectrum see Ref. [21]).
4,4,4-Trifluoro-3-(N-methylpyrrol-2-yl)butanoic acid (6a).
Colorless powder, 78% yield, Rf = 0.45.
1H NMR spectrum (DMSO-d6, δ, ppm, J/Hz): 2.81 (1H, d.d, 10.49 Hz, 16.03 Hz, CH(a)H(b)); 2.89 (1H, d.d, 4.45 Hz, 16.03 Hz, CH(а)Н(b)); H(a)/H(b) – AB system; 3.60 (3H, s, CH3); 4.02‑4.15 (1H, m, CHCF3); 5.96 (1H, m, Ar); 6.09 (1H, m, Ar); 6.72 (1H, m, Ar); 12.55 (1H, s, COOH).
19F NMR spectrum (DMSO-d6, δ, ppm, J/Hz): -69.82 (3F, br.s, CF3).
Mass spectrum (m/z (Irel (%)): 221 [M]+ (100), 201 [M-HF]+ (69), 162 [M-CH2COOH]+ (98), 142 [M-HF-CH2COOH]+ (12), 112 [M-CH2COOH-CF2]+ (37).
4,4,4-Trifluoro-3-(pyrrol-2-yl)butanoic acid (6b).
Colorless powder, 63% yield, Rf = 0.30.
1H NMR spectrum (DMSO-d6, δ, ppm, J/Hz): 2.81 (1H, d.d, 9.32 Hz, 16.23 Hz, CH(a)H(b)); 2.89 (1H, d.d, 5.37 Hz, 16.23 Hz, CH(а)Н(b)); H(a)/H(b) – AB system; 4.06-4.96 (1H, m, CHCF3); 5.99 (1H, m, Ar); 6.04 (1H, m, Ar); 6.72 (1H, m, Ar); 10.99 (1H, s, NH); 12.58 (1H, s, COOH).
19F NMR spectrum (DMSO-d6, δ, ppm, J/Hz): -69.95 (3F, s, CF3).
Mass spectrum (m/z (Irel (%)): 207 [M]+ (52), 187 [M-HF]+ (60), 148 [M-CH2COOH]+ (98), 128 [M‑HF-CH2COOH]+ (10).
4,4,4-Trifluoro-3-(5-methylpyrrol-2-yl)butanoic acid (6c).
Colorless powder, 75% yield, Rf = 0.45.
1H NMR spectrum (DMSO-d6, δ, ppm, J/Hz): 2.14 (3H, s, CH3); 2.78 (1H, d.d, 9.22 Hz, 16.05 Hz, CH(а)Н(b)); 2.86 (1H, d.d, 5.09 Hz, 16.05 Hz, CH(а)Н(b)); H(a)/H(b) – AB system; 3.82 -3.94 (1H, m, CHCF3); 5.65 (1H, m, Ar); 5.88 (1H, m, Ar); 10.69 (1H, s, NH); 12.49 (1H, s, COOH).
19F NMR spectrum (DMSO-d6, δ, ppm, J/Hz): -69.98 (3F, br.s, CF3).
Mass spectrum (m/z (Irel (%)): 221 [M]+ (41), 201 [M-HF]+ (81), 162 [M-CH2COOH]+ (62), 142 [M-HF-CH2COOH]+ (100).
Acknowledgements
This work was performed under financial support of the Ministry of Science and Higher Education of the Russian Federation in the framework of state task No. 075-00697-22-00 with the use of equipment of the Center for molecule composition studies of INEOS RAS.
References
- N. D. Chkanikov, A. S. Golubev, E. V. Belyaeva, INEOS OPEN, 2019, 2, 33; DOI: 10.32931/io1906r.
- G. Sandford., I. Wilson, C. M. Timperley, J. Fluor. Chem., 2004, 125, 1425; DOI: 10.1016/j.jfluchem.2004.04.013.
- C. O. Okoro, O. O. Fadeyi, P. L. Jackson, R. L. Richmond., T. Farmer, Tetrahedron Lett., 2006, 47, 7451; DOI: 10.1016/j.tetlet.2006.08.046.
- A.Yu. Volkonskii, E. M. Kagramanova, N. D. Kagramanov, N. D. Chkanikov, Russ. Chem. Bull., 2012, 61, 2001; DOI: 10.1007/s11172-012-0278-0.
- B. Guo, D. S. Zijlstra, J. G. de Vries, E. Otten, ChemCatChem., 2018, 10, 2868; DOI: 10.1002/cctc.201800509.
- X. Li, Y. Qi, R Qi, D. Bai, Tetrahedron Lett., 2020, 61, 151858; DOI: 10.1016/j.tetlet.2020.151858.
- O. Yu. Fedorovskii, A. Yu. Volkonskii, A. S. Golubev, Yu. Ya. Spiridonov, N. D. Chkanikov, Russ. Chem. Bull., 2017, 66, 1116; DOI: 10.1007/s11172-017-1863-z.
- A. V. Fokin, V. Yu. Tyutin, N. D. Chkanikov, Russ. Chem. Rev., 1992, 61, 766; DOI: 10.1070/RC1992v061n08ABEH000997.
- N. D. Chkanikov., K. V. Komarov, V. Yu. Tyutin, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1991, 40, 1070; DOI: 10.1007/BF00961377.
- N. D. Chkanikov, O. Yu. Fedorovskii, Fluorine Notes, 2021, 134, 3; DOI: 10.17677/Fn20714807.2021.01.02.
- N. D. Сhkanikov, O. Yu. Fedorovskii, Fluorine Notes, 2022, 140, 3; DOI: 10.17677/fn20714807.2022.01.02.
- G. A. Olah, R. Krishnamurti, G. K. S. Prakash, Comprehensive Organic Synthesis, Elsevier, 1991, 7, 293; DOI: 10.1016/B978-0-08-052349-1.00065-2.
- A. Berlin, A. Canavesi, G. Schiavon, S. Zecchin, G, Zotti, Tetrahedron, 1996, 52, 7947; DOI: 10.1016/0040-4020(96)00366-3.
- B. Pal, V. S. Giri, P. Jaisankar, Catal. Commun., 2005, 6, 711; DOI: 10.1016/j.catcom.2005.07.003.
- T. Dohi, K. Morimoto, M. Ito, Y. Kita, Synthesis (Stuttg), 2007, 18, 2913; DOI: 10.1055/s-2007-983798.
- I. D. Gridnev, N. A. Gridneva, Russ. Chem. Rev., 1995, 64, 1021; DOI: 10.1070/RC1995v064n11ABEH000191.
- P. L. Coe, A. G. Holton, G. C. Tatlow, J. Fluor. Chem., 1982, 21, 171; DOI: 10.1016/S0022-1139(00)81240-3.
- E. N. Zilberman, Reactions of Nitriles, Khimia, Moscow, 1972 (in Russian).
- JP 05279331; Chem. Abstrs., 1994, 120, 163976h.
- M. Katayama, R. K. Gautam, J. Pesticide Sci., 1997, 22, 331; DOI: 10.1585/jpestics.22.331.
- M. Katayama, R. K. Gautam, Biosci. Biotech. Biochem., 1996, 60, 755; DOI: 10.1271/bbb.60755.
ARTICLE INFO
Received 27 June 2023
Accepted 20 July 2023
Available online August 2023
Recommended for publication by PhD O.V. Bryzgalova
eLIBRARY Document Number (EDN) KVHZHG

Fluorine Notes, 2023, 149, 1-2
