Received: August 2022
DOI 10.17677/fn20714807.2022.04.01
Fluorine Notes, 2022, 143, 1-2
IODINE(III) TRIS(TRIFLUOROACETATE) IN REACTIONS OF IODO- AND BROMOTRIFLUOROACETOXYLATION OF FLUOROOLEFINS
A.A. Tyutyunov
A.N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences,
28 Vavilova St., 119991 Moscow, Russian Federation
E-mail: tuytuynov@rambler.ru
Abstract: Reactions of iodine (III) tris(trifluoroacetate) with perfluoroalkylethylenes and fluoroolefins were studied. The reaction of I(OCOCF3)3 with ethylene derivatives bearing CF3-, C4F9-, and sterically demanding C3F7C(CF3)2 moieties gives the corresponding products of iodotrifluoroacetoxylation. In turn, the reactions involving tetrafluoroethylene and 1,1-difluoroethylene afford a mixture of fluoroanhydrides, which are further converted into CF3COF.
Keywords: iodine(III) tris(trifluoroacetate), iodotrifluoroacetoxylation, fluoroolefins, 1,1,1‑trifluoro-2,3-epoxypropane.
The synthetic applications of polyvalent iodine derivatives enable the development of effective approaches to various organic and particularly organofluorine compounds [1-3]. Along with iodine (III) derivatives used as perfluoroalkylating reagents, such as FITS reagents [4], Togni reagent [5] and organoiodine(V) derivatives, e.g., Dess-Martin periodinane (DMP) [6] applied for oxidation, reactions of organofluorine compounds with interhalides [7], as well as with inorganic sulfonates and sulfates of polyvalent iodine [8], have been studied in detail. The purpose of the present study was to investigate the possibility of using iodine (III) tris(trifluoroacetate) [9-10] in iodotrifluoroacetoxylation of perfluoroalkylethylenes and fluoroolefins.
Previously it was shown that iodine (III) tris(trifluoroacetate) reacts with a variety of olefins to give vicinal diol trifluoroacetates [11]. This reaction was found to occur via iodotrifluoroacetoxylation of an alkene followed by the replacement of iodine with trifluoroacetate anion [12-15].
It turned out that perfluoroalkylethylenes 1a-d react with iodine (III) tris(trifluoroacetate) in the presence of an equivalent amount of iodine already at 15-25°С to afford iodotrifluoroacetoxylation products 2a-d in high yields. Ethylene derivatives bearing perfluoroalkyl substituents of various lengths and steric bulk enter this reaction. However, alkene 1d, containing the sterically demanding perfluoro-tert-hexyl substituent, reacts with I(OCOCF3)3 only on a prolonged heating of the reaction mixture to a temperature of about 50С. It is more convenient to convert the reaction products 2a-d to the corresponding iodohydrins 3a-d prior to isolation, since iodotrifluoroacetates 2a-d partially hydrolyze during an aqueous work-up of the reaction mixture.
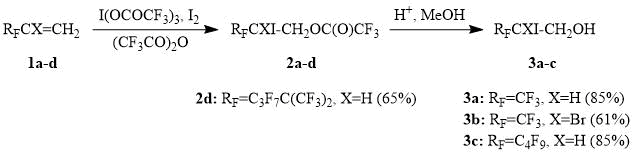
Scheme 1.
Iodotrifluoroacetylation of perfluoroalkylethylenes could be implemented for the preparative synthesis of perfluoroalkyloxiranes, as demonstrated by the preparation of (trifluoromethyl)oxirane, a valuable organofluorine reagent used in the synthesis of various trifluoromethyl-containing products and polymers [16]. It is worth noting that in this case the reaction of iodine (III) tris(trifluoroacetate) with 3,3,3-trifluoropropene is carried out in the presence of bromine and furnishes a mixture of iodo- and bromotrifluoroacetoxylated products, which reduces the consumption of iodine, which is significantly more expensive than bromine.
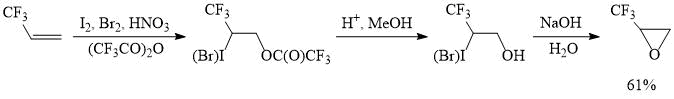
Scheme 2.
Studies of the reactions of various fluoroolefins with iodine (III) tris(trifluoroacetate) showed that tetrafluoroethylene and 1,1-difluoroethylene react with I(OCOCF3)3. In turn, hexafluoropropene and perfluorocyclohexene do not react with it at temperatures of 20-50°С.
It has recently been shown that oxidation of iodine with fuming nitric acid according to the method [9] in a mixture of CF3CO2H/(CF3CO)2O or pure trifluoroacetic anhydride produces a nitrosonium dinuclear trifluoroacetate iodine (III) complex with one bridging μ-CF3СO2- ligand [17]. In this case, the 13C NMR spectrum of the reaction mixture show two signals at 113 (q, 1JCF = 283 Hz, CF3) and 162 (q, 2JCF = 42 Hz, CF3CO2) ppm, which correspond to trifluoroacetate anion in [I(OCOCF3)3]2(CF3CO2)NO [17-18]. After adding an equivalent amount of iodine to the reaction mixture and slow bubbling of tetrafluoroethylene, which is initially absorbed, after a few hours the gas begins to evolve from the reaction mixture. As a result, the reaction mixture is almost completely evaporated to give iodine, and the evolved gas condenses in a trap cooled to -78C as a dark blue liquid. We have found that when methanol is slowly and carefully added to the condensed gas, CF3CO2Me is produced (13С NMR, δ: 115 (q, 1JCF = 283 Hz, CF3), 158 (q, 2JCF = 42 Hz, CF3CO2)), while products bearing the ICF2 functionality are formed only in trace amounts (as is evident from the 13C and 19F NMR spectra).
A similar reaction using an equivalent amount of bromine in place of iodine occurs in the same way. However, in this case, after the absorption of tetrafluoroethylene and the completion of gas evolution, in addition to iodine, a liquid residue remains, the analysis of which by 13С NMR spectroscopy showed that beside signals corresponding to the CF3CO2- anion, the signals of BrCF2CO2H appear (13С NMR, δ: 107 (t, 1JCF = 312 Hz, BrCF2), 165 (t, 2JCF = 31 Hz, BrCF2CO2)), and according to the 19F NMR data, there are no acid fluorides in the reaction mixture and only signals corresponding to trifluoroacetic and bromodifluoroacetic acids are observed. The evolving gas contains mainly CF3COF with a minor admixture of BrCF2COF, with no traces of 1,2-dibromotetrafluoroethane – BrCF2CF2Br (13С NMR, δ: 114, tt, 1JCF = 310 Hz, 2JCF = 39 Hz) being detected among the reaction products.
Therefore, based on experimental data, it can be assumed that the reaction of tetrafluoroethylene with I(OCOCF3)3 in the presence of I2 (1 equiv.) gives primarily the product of iodotrifluoroacetoxylation, ICF2CF2OCOCF3, which is an unstable perfluorinated ester that decomposes into two acid fluorides, CF3COF and ICF2COF, the first of which escapes from the reaction mixture in the form of a gas (CF3COF, b.p. -59C), carrying with it a small amount of ICF2COF (b.p. 40оC, 13С NMR, δ: 79 (td, 1JCF = 320 Hz, 2JCF = 89 Hz, ICF2), 148 (dt, 1JCF = 368 Hz, 2JCF = 34 Hz, ICF2COF); 19F NMR, δ: -61.3 (d, 3JFF = 8 Hz, ICF2), 7 (t, 3JFF = 8 Hz, COF)), while the second one, ICF2COF, decomposes in the reaction mixture to yield, probably, I2+CF3COF+(COF)2. In turn, the reaction of tetrafluoroethylene with a I(OCOCF3)3 + Br2 (1 equiv.) primarily produces a mixture of iodo- and bromotrifluoroacetoxylation products, ICF2CF2OCOCF3 + BrCF2CF2OCOCF3, which decompose to afford three acid fluorides: CF3COF, BrCF2COF and ICF2COF, the latter from which decomposes in the above-mentioned way, and BrCF2COF reacts with trifluoroacetic acid, presenting in the reaction mixture, to give BrCF2CO2H and CF3COF. At the same time, it is evident from these experiments that the BrCF2CO moiety is stable under the reaction conditions and avoid decomposition.
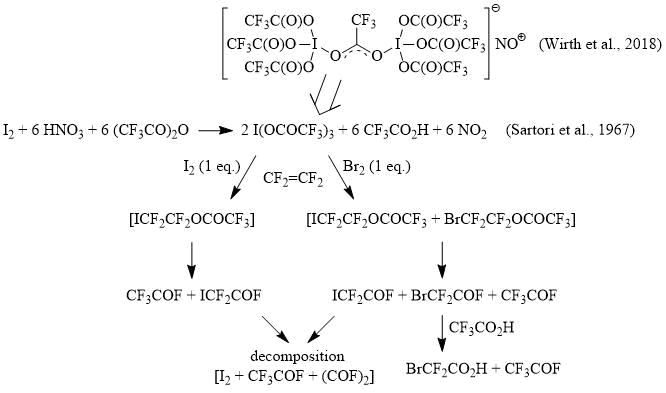
Scheme 3
The reaction of iodine (III) tri(trifluoroacetate) with 1,1-difluoroethylene was carried out in a similar way and is also accompanied by the transformation of the fluoroolefin (mainly into CF3COF, which has no significant practical interest.
To conclude, as a result of our research, it was shown that iodine (III) tris(trifluoroacetate) can be applied to convert perfluoroalkylethylenes into iodotrifluoroacetoxylation products, which can then be used to obtain the corresponding oxiranes.
Experimental
1H, 13C and 19F NMR spectra were recorded on a Bruker AVANCE-400 and Bruker AM-300 spectrometers with 400, 100 and 282 MHz working frequencies, respectively, using CDCl3 as an external standard. Chemical shifts in the 1H NMR spectra were referenced to the residual proton chloroform peak (7.26 ppm) and reported in ppm units relative to TMS. Chemical shifts in the 13C NMR spectra were referenced to the 13С nucleus peak (77.0 ppm in CDCl3) and reported in ppm units relative to TMS. Chemical shifts in 19F NMR spectra were reported in ppm units relative to an external CFCl3 reference. Positive values of chemical shift correspond to the downfield shift of the indicator nucleus signal.
Iodotrifluoroacetoxylation of perfluoroalkylethylenes (general procedure)
To a stirred mixture of granulated iodine (8.6 g, 0.0338 mol) in trifluoroacetic anhydride (30 mL) at 10÷15°С, fuming nitric acid (9 mL, 0.214 mol, d=1.5 g cm-3) is added dropwise, during which a slight gas release and a dissolution of iodine are observed. More granulated iodine (17.2 g, 0.0677 mol) is then added to a resulting stirred clear, light-brown solution, followed by the addition of perfluoroalkylethylene 1a-d (0.2 mol) at 15÷20°С. After this, the reaction mixture is stirred for 1 h at 25С, poured onto an ice-water mixture, the lower layer is washed with cold water, aqueous solutions of sodium sulfite and sodium chloride.
The product of iodotrifluoroacetoxylation of perfluoroalkylethylene 2a-d is obtained in admixture with the corresponding iodohydrine 3a-d.
The product is dissolved in methanol (15 mL), 50% aqueous solution of sulfuric acid (1 mL) is added, and the resulting mixture is heated with a distillation column, distilling off methyl trifluoroacetates. Next, the reaction mixture is poured into the cold sodium chloride solution, the lower layer is separated and the product 3a-c is purified by vacuum distillation and/or recrystallization.
2-Iodo-3,3,3-trifluoropropan-1-ol (3a).
Yield 40 g (85%). B.p. 57°С (20 Torr). Found (%): C, 15.01; H, 1.51; F, 23.65. C3H4F3IO. Calculated (%): C, 15.02; H, 1.68; F, 23.75. 1H NMR, δ: 3.55 (m, 2H, CH2), 4.05 (m, 1H, CHI), 4.2 (s, 1H, OH); 19F NMR, δ: -68.1 (d, 3JFH = 5.5 Hz, CF3).
2-Iodo-2-bromo-3,3,3-trifluoropropan-1-ol (3b).
Yield 38.9 g (61%). B.p. 81-82°С (20 Torr), m.p. 60C. Found (%): C, 11.16; H, 0.71; F, 17.77. C3H3BrF3IO. Calculated (%): C, 11.30; H, 0.95; F, 17.87. 1H NMR, δ: 3.7 (s, 1H, OH), 4.02, 4.08 (AB q, 2H, 2Jab = 13 Hz, CH2); 19F NMR, δ: -71.6 (s, 3F, CF3).
2-Iodo-3,3,4,4,5,5,6,6,6-nonafluorohexan-1-ol (3c).
Yield 66 g (85%). B.p. 117°С (20 Torr). Found (%): C, 18.48; H, 1.03; F, 43.90. C6H4F9IO. Calculated (%): C, 18.48; H, 1.03; F, 43.84. 1H NMR, δ: 2.50 (s, 1H, OH), 3.97 (d, 2H, 3JHH = 6 Hz, CH2), 4.59 (m, 1H, CHI); 19F NMR, δ: -127.3, -126.4 (AB q, 2F, 2Jab = 288 Hz, CF2), -121.6, -120.5 (AB q, 2F, 2Jab = 296 Hz, CF2), -111.5, -98.4 (AB q, 2F, 2Jab = 277 Hz, CF2), -81.9 (s, 3F, CF3).
2-Iodo-3,3-bis(trifluoromethyl)-4,4,5,5,6,6,6-heptafluorohexyl trifluoroacetate (2d).
After the addition of olefine 1d, the reaction mixture was heated to reflux for 5–6 h and isolated 2d.
Yield 76 g (65%). B.p. 50°С (0.5 Torr). Found (%): C, 20.01; H, 0.70; F, 52.07. C10H3F16IO2. Calculated (%): C, 20.50; H, 0.52; F, 51.87. 1H NMR, δ: 4.7 (m, 2H, CH2), 4.75 (m, 1H, CHI); 19F NMR, δ: -124.5 (m, 2F, CF2), -106, -103.5 (AB q, 2F, 2Jab = 319 Hz, CF2), -82.7 (m, 3F, CF3CF2), -77.7 (s, 3F, C(O)CF3), -62.1 (m, 3F, C(CF3)), -61.5 (m, 3F, C(CF3)).
1,1,1-Trifluoro-2,3-epoxypropane.
A 6 L flask was charged with trifluoroacetic acid anhydride (2 L), iodine (430 g, 1.69 mol) and then, fuming nitric acid (450 mL, d=1.5 g cm-3, 10.7 mol) was added at 10-15°С. The resulting mixture was stirred at 20-25°С for 2 h, cooled to 15°С, after which bromine (542 g, 3.39 mol) was added, stirred for 15 min and gaseous trifluoropropene (1 kg, 10.4 mol) was introduced, while maintaining the temperature at 15-20°С. Then, the mixture was stirred for one h at 23-28°С and poured onto a crushed ice-water mixture. The lower layer was separated, washed with an equal volume of water, aqueous solutions of sodium sulfite and sodium chloride to afford a mixture (~2.8 kg) of iodo- and bromotrifluoroacetoxylation products in a ratio of 1:2 with a small admixture of the corresponding halohydrines. This mixture was used without further purification.
The obtained product was charged in a 6 L flask, dissolved in a mixture of methanol (1.5 L) and 50% sulfuric acid (50 mL), and heated with distillation (with a reflux condenser or a column of 20-25 cm) of methyl trifluoroacetate, then the temperature was raised and the remaining methanol was distilled off; the mixture was heated to 90-95C until the distillation of methanol stops. After this, the mixture was cooled to 80-85°С and aqueous solution of NaOH (480 g of NaOH in 2L of water) was added, so that the newly formed reaction product is distilled off not too rapidly. The resulting distillate was washed with an equal volume of ice water to give a crude product (888 g, 90% purity), which was dried and distilled to afford (trifluoromethyl)oxirane (700 g, 97% purity).
Acknowledgements
This study was performed within the State task of the Ministry of Science and Higher Education of the Russian Federation, Project No. 075-00697-22-00.
References
- V.V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley & Sons, Ltd, ISBN: 9781118341032, 2014.
- A. Yoshimura, V.V. Zhdankin, Chem. Rev., 2016, 116, 3328-3435.
- Hypervalent Iodine Chemistry, Editor T. Wirth, Top. Curr. Chem., 373, Springer, ISBN: 9783319337319, 2016.
- T. Umemoto, Chem.Rev., 1996, 96, 1757-1778.
- P. Eisenberger, S. Gischig, A. Togni, Chem. Eur. J., 2006, 12, 2579-2586.
- V.V. Zhdankin, J. Org. Chem., 2011, 76, 1185-1197.
- R.D. Chambers, W.K.R. Musqrave, J. Savory, J. Chem. Soc., 1961, 3779-3786.
- T.M. Kasumov, A.S. Koz’min, N.S. Zefirov, Russ .Chem. Rev., 1997, 66, 843-857.
- M. Schmeisser, K. Dahmen, P. Sartori, Chem. Ber., 1967, 100, 1633-1637.
- R.M. Moriarty, J.W. Kosmeder II, V. Sikervar, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2014, https://doi.org/10.1002/047084289X.ri024.pub2
- J. Buddrus, Angew. Chem. Int. Ed., 1973, 12, 163-164.
- J. Buddrus, H. Plettenberg Chem.Ber., 1980, 113, 1494-1506.
- F. Cech, M. Linskeseder, E. Zbiral, Monatshef. Chem., 1976, 107, 1429-1436.
- M. Linskeseder, E. Zbiral, Liebigs Ann. Chem., 1977, 1039-1049.
- R.C. Cambie, D. Chambers, P.S. Rutledqe, P.D. Woodgate, J. Chem. Soc., Perkin Trans. 1, 1977, 2231-2235.
- P.V. Ramachandran, K.J. Padiya, JFC, 2007, 128, 1255-1259.
- T. Hokamp, L. Mollari, L.C. Wilkins, R.L. Melen, T. Wirth, Angew. Chem. Int. Ed., 2018, 57, 8306-8309.
- K. Matsuoka, N. Komami, M. Kojima, T. Mita, K. Suzuki, S. Maeda, T. Yoshino, S. Matsunaga, J. Am. Chem. Soc., 2021, 143, 103-108.
ARTICLE INFO
Received 08 August 2022
Accepted 16 August 2022
Available online August 2022
Recommended for publication by PhD M. Manaenkova
eLIBRARY Document Number (EDN) SKKOFT

Fluorine Notes, 2022, 143, 1-2
