Received: December 2022
DOI 10.17677/fn20714807.2022.01.04
Fluorine Notes, 2022, 140, 7-8
DECARBOXYLATION OF 2,3,3,3-TETRAFLUORO-2-IODOPROPIONIC ACID, ITS POTASSIUM SALT AND TRIMETHYLSILYL ESTER
V.E. Boikoab, A.V. Edunovb, S.М. Igumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences,28 Vavilova St., 119991 Moscow, Russia.
bP@M-Invest Scientific Production Association, 119991, Leninsky ave. 47, Moscow, Russia.
e-mail: boykii@mail.ru
Abstract: The preparation of 1,1,1,2-tetrafluoro2-iodoethane by various methods has been studied, it has been shown that the best method is a method in which a solution of acid in dimethylformamide is gradually added to dimethylformamide preheated to 140°C, the mechanism of this reaction with the formation of CF3CFI – anion is proposed.
Keywords: iodofluoroalkanes, decarboxylation, 2,3,3,3-tetrafluoro2-iodopropionic acid.
Iodofluoroalkanes are the basic synthones in organofluorine chemistry; they are widely used for fluoroalkylation of aliphatic, aromatic, heterocyclic, organoelement compounds. [1-3]. Lower iodofluoroalkanes are also used as telogens for telomerisation to form products, used for the synthesis of fluoroacids [4], fluorinated alcohols [5]; monomers for thermo- and chemically stable polymers [6].
We have recently proposed an original method for obtaining pentafluoroethyliodide from perfluoropropylene [7], the key intermediate in which is the fluorohydride 2,3,3,3-tetrafluoro2-iodopropionic acid I. In continuation of this work, it was proposed to obtain 1,1,1,2-tetrafluoro2-iodoethane and 1,1,2-trifluoro2-iodoethene using the same intermediate compound.
It is known to produce 1,1,1,2-tetrafluoro-2-iodoethane by the decarboxylation of tetrafluoropropionic acid salts in the presence of iodine in dimethylformamide with the yield about 30 % [8], through the addition of HF to 1-iodo-1,2,2,2-tetrafluoroethane in gas phase on the chrome catalyst [9], through the iodofluorination of 1,1,2-trifluoroethene by the mixture of iodine and IF5 using aluminum with aluminium iodide as catalysts at temperatures 130-140C in an autoclave [10], and also through the reaction of 1,1,2-trifluoroethene with ICl in HF/BF3 medium at 25C with a yield 73 % [11].
The aim of this work was to obtain 1,1,1,2-tetrafluluoro-2-iodoethane and 1,1,2-trifluoro-2-iodoethene from 2,3,3,3-tetrafluoro-2-iodopropionic acid II and its trimethylsilyl ester III, which, in turn, were obtained from fluoro-anhydride I according to the Scheme 1 below.
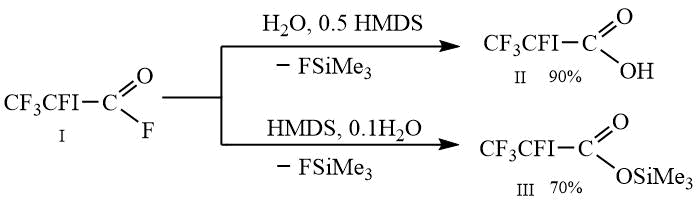
Scheme 1.
Depending on the conditions of decarboxylation of acid II (Scheme 2), we obtained different results. During the decarboxylation process with simultaneous distillation of the resulting volatile products, the yield of 1,1,1,2-tetrafluoro2-iodoethane I was 48% (Method 1). In the case of using the method consisting in the gradual addition of an acid solution in dimethylformamide to dimethylformamide preheated to 140°C (Method 2), it was possible to increase the yield of the desired product IV to 72%. However, carrying out the reaction without simultaneous distillation of volatile products (Method 3) led to an unexpected result. After isolation and comparison of GC data, 1H, 19F NMR spectra and mass spectra, it turned out that the reaction products in addition to IV are 1,1,1,2-tetrafluoro-2,2-diiodethane V and 2,3,3,3-tetrafluoropropionic acid VI.
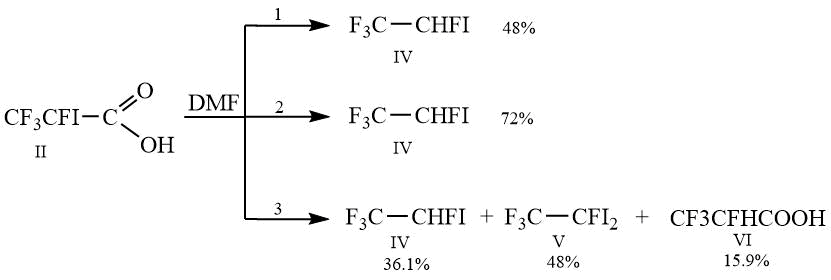
Scheme 2.
Decarboxylation of silyl ether III did not lead to the formation of the target 1,1,2-trifluoro-2-iodoethene VII and trimethyl(1,2,2,2-tetrafluoro1-iodoethyl)silane IX, the only reaction product was 1,1,1,2-tetrafluoro-2,2-diiodethane V.
As an alternative to obtaining 1,1,2-trifluoro2-iodoethene VIII, decarboxylation of the potassium salt 2,3,3,3-tetrafluoro2-iodopropionic acid was carried out, however, in this case the formation of the expected products did not occur, the reaction resulted in compounds IV and V in a ratio of 1:3.
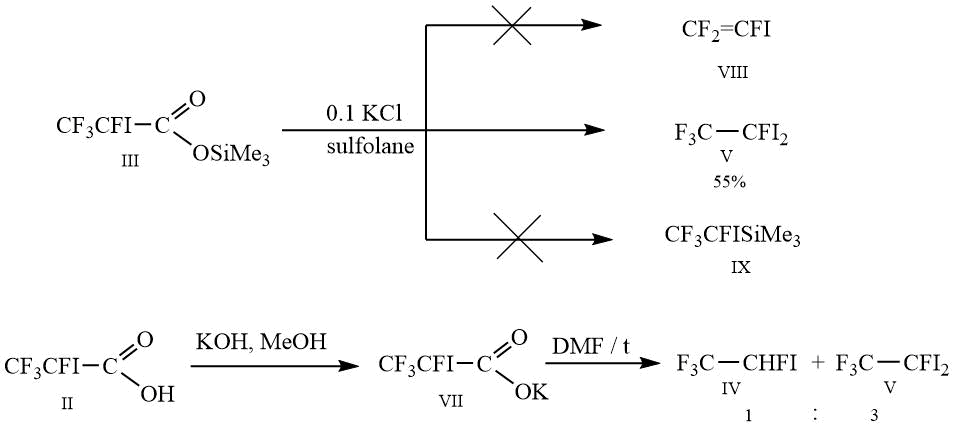
Scheme 3.
By analyzing the obtained experimental data, it can be assumed that, as a result of decarboxylation, a stable CF3CFIanion is formed, which is reacted as shown in Schemes 4 and 5.
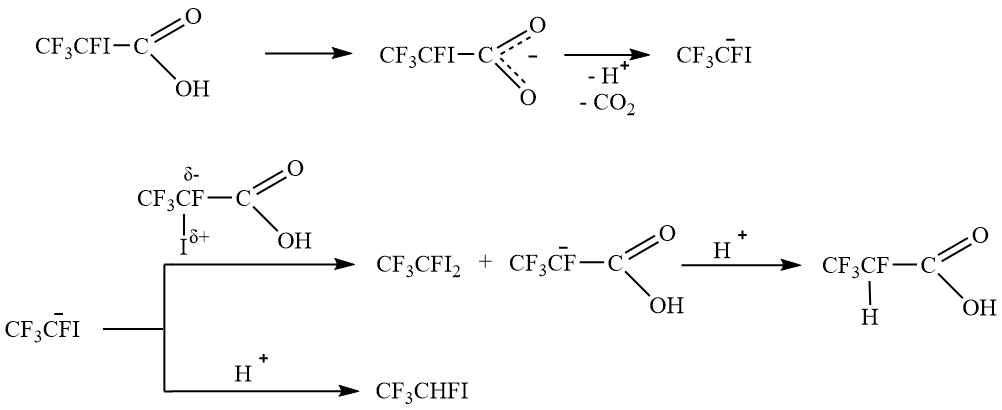
Scheme 4.
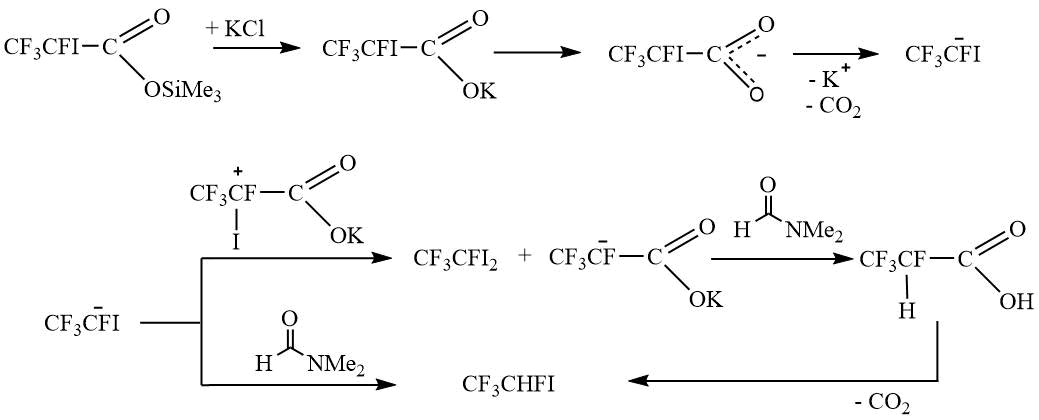
Scheme 5.
Thus, the formation of the diiodo derivative V, and not assumed alkene VIII or trimethylsilane IX, predominantly occurs.
Experimental
1H and 19F NMR spectra were recorded on the “Bruker AVANGE-300” spectrometer at 300 and 282 MHz correspondingly, external standard is CDCl3. Chemical shifts for 1H NMR spectra are given in ppm relative to TMS. Chemical shifts for 19F NMR spectra are given in ppm relative to CFCl3.
Fluoro-ahydride I was obtained as described in [7].
Preparation of 2,3,3,3-tetrafluoro-2-iodopropionic acid II
To 6.9 g (0.383 mol) of water and 31.1 g (0.192 mol) of hexamethyldisilane, while stirring, 105 g (0.383 mol) of fluoro-anhydride I is added dropwise. While adding fluoro-anhydride, the temperature rises to 34C and the formation of trimethylsilane is observed. The reaction mass is kept at this temperature for two hours, then 15 g of fluorotrimethylsilane is distilled in vacuum into a receiving flask cooled by a mixture of dry ice with acetone. Next, the receiving flask is replaced, 0.2 g of copper powder is added to the cube and 94 g of 2,3,3,3-tetrafluoro2-iodopropionic acid II is distilled as a colorless liquid. Yield 90 %, purity is 97 %, m.p. 28-31C, b.p. 67-690C/20 mm Hg.
1H NMR, δ, ppm: 10.1 (с, 1H, CООН);
19F NMR, δ, ppm: -142.3 (m, 1F, CFI), -76.5 (m, 3F, CF3).
Preparation of trimethylsilyl ester of 2,3,3,3-tetrafluoro-2-iodopropionic acid III
To 0.7 g (0.0394 mol) of water and 64 g (0.394 mol) of hexamethyldisilane, while stirring, 108g (0.394 mol) of fluoro-anhydride I is added dropwise. While adding of fluoro-anhydride the temperature rises to 46C and the formation of trimethylfluorosilane is observed. The reaction mass is heated to 62C and kept by this temperature during 1 hour, then 10 g of the mixture of fluorotrimethylsilane and hexamethyldisilane are distilled into receiving flask which is cooled by mixture of dry ice and acetone. Further the receiving flask is replaced, 0.2 g of copper powder are added into the still and 95 g of 2,3,3,3-tetrafluoro-2-iodopropionic acid III are distilled as colorless liquid. Yield 70 %, purity 97 % by GC, b.p/ 52-53C/20 mm Hg
1H NMR, δ, ppm: 0.4 (с, 9Н SiMe3);
19F NMR, δ, ppm: -138.6 (m, 1F, CFI), -76.8 (m, 3F, CF3).
Preparation of 1,1,1,2-tetrafluoro-2-iodoethane IV
Method 1.
The solution of 20 g (0.0735 mol) II in 20 g of DMF, while stirring, is gradually heated to the temperature 135-137C, by which gas emission begins. Then the reaction mixture is slowly heated to 145C, and 1,1,1,2-tetrafluoro-2-iodoethane is simultaneously distilled into receiving flask which is cooled by mixture of dry ice and acetone. Yield is 48 %, purity 97 %, b.p. 39C.
Preparation of 1,1,1,2-tetrafluoro-2-iodoethane IV
Method 2.
To 20 g of DMF, which is heated to 140C, while stirring 20 g (0.0735 mol) of II is added dropwise into 15 g of DMF and 12 g of 1,1,1,2-tetrafluoro-2-iodoethane is simultaneously distilled into receiving flask which is cooled by mixture of dry ice and acetone. Yield i72 %, purity 97%, b.p. 39C.
Preparation of 1,1,1,2-tetrafluoro-2-iodoethane IV
Method 3.
The solution of 20 g (0.0735 mol) II in 20 g of DMF is loaded into a flask, provided with reflux, which is connected to deep-cooling condenser, and while stirring, gradually heated to 140C. It is kept by this temperature during 30 minutes upon the completion of gas emission, then the reaction mass is cooled to room temperature and poured into double volume of 15% hydrochloric acid. The lower layer is separated and washed by 2х15 ml of water, dried over CaCl2. It is obtained 17 g of mixture, consisting according to GC from 36.1 % IV, 48% V, 15.9 % VI.
Preparation of 1,1,1,2-tetrafluoro-2,2-diiodoethane V
The solution of 20 g (0.0579 mol) of trimethylsilyl ester III and 0.4 g of potassium chloride (0.00579 mol) in 20 g of absolute sulfolane, while stirring, is gradually heated to the temperature 90-93C, by which gas emission begins. Then the reaction mixture is gradually heated to 150C, and 5 g of trimethylfluorosilane are simultaneously distilled into receiving flask, which is cooled by mixture of dry ice and acetone. The reaction mass is cooled to room temperature and poured into double volume of water, washed by 2х15 ml of water, dried over CaCl2 and distilled. It is obtained 6.5 g of 1,1,1,2-tetrafluoro-2,2-diiodoethane, purity 96 %, b.p. 128-130C.
19F NMR, δ, ppm: -97.7 (m, 1F, CFI2), -80.6 (m, 3F, CF3).
Preparation of potassium salt of 2,3,3,3-tetrafluoro-2-iodopropionic acid VII
To the solution of 10 g (0.0367 mol) of acid II in 15 ml of methanol, while stirring, 2 g (0.0367 mol) of potassium hydroxide in 7 ml of methanol is added dropwise. It is stirred during 20 minutes, methanol is distilled in vacuum of water-jet pump, the salt is dried over P2O5. The yield is quantitative.
Decarboxylation of potassium salt of 2,3,3,3-tetrafluoro-2-iodopropionic acid
In a 100 ml two-neck flask, equipped with thermometer and reflux condenser, which is connected to deep-cooling condenser, cooling by the mixture dry ice-acetone, with Tishchenko flask with concentrated H2SO4, 11 g (0.0355 mol) of VII in 16 g of DMF are loaded, the solution, while stirring, is gradually heated to 93C and stirred by this temperature during 30 minutes upon the completion of gas emission. The reaction gas is cooled to room temperature and poured into double volume of 15 % hydrochloric acid, washed by 2х5 ml of water, dried over CaCl2. It is obtained 3.2 g of crude product, containing according to GC: 26 % IV, 74 % V.
Acknowledgments
This work was performed with the financial support from Ministry of Science and Higher Education of the Russian Federation. The contribution of Center for molecular composition studies of A.N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences (Moscow, Russia) is gratefully acknowledged.
References
- Tiers G., Journal of the American Chemical Society, 1960, 82(20), 5513.
- Patent US 3271441 (1966).
- V.C.R. McLoughlin, J.Thrower, Tetrahedron, 1969, 25(24), 5921-5940.
- Patent US 3351644 (1967).
- Patent US 3576885 (1971).
- Patent US 3862077 (1975).
- V.E. Boiko, A.V. Edunov, S.М. Igumnov, Fluorine notes, 2016, 6(109), 5-6.
- "Syntheses of organofluorine compounds", Part 3, edited by S.M. Igumnov, E.V. Igumnova, M., "Trovant", 2015, 214p., 3. (in Russian)
- Patent DE 3009760 (1981).
- Hauptschein M., Braid M., JACS, 1961, 83, 2383-2386.
- Petrov V.A., Krespan C.G., J. Org. Chem., 1996, 61, 9605-9607.
ARTICLE INFO
Received 09 December 2021
Accepted 23 December 2021
Available online February 2022
Recommended for publication by PhD M. A. Manaenkova
Fluorine Notes, 2022, 140, 7-8
