Received: February 2022
DOI 10.17677/fn20714807.2022.01.02
Fluorine Notes, 2022, 140, 3-4
C2-Alkylation in a three-component reaction of fluorocarbonyl compounds, pyrrole derivatives and malononitrile in competition with C2-oxyalkylation
O.Yu. Fedorovskii, N.D. Chkanikov
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences,
Vavilova 28, Moscow,
119991 Russia (offskii@rambler.ru)
Abstract: The C2-alkylation of pyrrole derivatives with 2-(2,2,2-trifluoroethylidene) malononitrile 1, formed in situ from ethyl hemiacetal trifluoroacetaldehyde 2 and malononitrile in the presence of a base, has been carried out. As a result of this three-component reaction, nitriles were obtained - derivatives of 4,5,6,7-tetrahydroindole and pyrrole 4a, b, c. A competing reaction is C2-oxyalkylation of the pyrrole ring with hemiacetal 2. It has been shown that the side reaction can be completely suppressed by lowering the reaction temperature to -30°C. When the ethyl ester of trifluoropyruvic acid 3 reacts with pyrrole derivatives in the presence of malononitrile, the main reaction is the C2-oxyalkylation of the pyrrole ring, rather than its C2-alkylation formed in situ by alkene 10. Nitriles 6 - 9 were obtained in high yields from pyrrole derivatives and alkene 10. The structure of the compounds was confirmed by 1H, 13C, 19F NMR spectroscopy, mass spectrometry and high-resolution mass spectrometry.
Keywords: 2-(2,2,2-trifluoroethylidene)malononitrile, ethyl 3,3-dicyano-2-(trifluoromethyl) acrylate, substitution of 2-(2,2,2-trifluoro-1-(1H-pyrrol-2-yl)ethyl)malononitrile and ethyl 2-(dicyanomethyl)-3,3,3-trifluoro-2-(4,5,6,7-tetrahydro-1H-indol-2-yl)propanoate, three-component one-step synthesis, C2-alkylation, C2-oxyalkylation, trifluoroacetaldehyde ethyl hemiacetal, ethyl trifluoropyruvate, pyrrole and indole derivatives, malononitrile.
Introduction
The synthesis of heterocyclic organofluorine compounds has attracted a lot of attention for many years. Their promise is especially noticeable in pharmaceutical chemistry; about 20% of all drugs contain at least one fluorine atom. However, the synthesis of these compounds often causes unexpected difficulties [1-4]. Trifluoroacetaldehyde ethyl hemiacetal 2 and trifluoropyruvic acid ethyl ester 3 are some of the most important synthones with which heterocyclic compounds containing a trifluoromethyl group can be prepared. We previously reported on a three-component reaction of indole derivatives, 2 and malononitrile in the presence of a base, leading to 3-(1H-indol-3-yl)-2-cyano-4,4,4-trifluorobutanoic acid nitriles in good yields [4]. Cyano pyrrole derivatives and THI are potentially biologically active substances, and can also be used as intermediates in further directed synthesis to search for new biologically active compounds or plant growth regulators [5]. Currently, multicomponent reactions have reached the level of advanced synthetic tools, the search and development of which is stimulated by the principles of waste-free chemical production, that is, "green chemistry". The use of the three-component synthesis procedure allows avoiding the stage of obtaining, purifying, and using fluorine-containing cyanoethylenes, which are known for their toxicity [6, 7].
In this work, we applied a three-component one-step synthesis to obtain fluorine-containing cyanide derivatives of pyrrole and indole, substituted at position 2, based on fluorocarbonyl compounds 2 and 3.
Results and discussion
Developing systematic studies of the synthesis of fluorine-containing heterocyclic compounds, starting
from fluorine-containing cyanoethylenes, we showed that the C2-alkylation of indoles
with such a reactive alkene as 2-(2,2,2-trifluoroethylidene)malononitrile 1 can be carried out as a result of the three-component reaction of indoles, hemiacetal 2 and malononitrile in the presence of a base, i.e. without isolation of alkene formed in situ [4] (Scheme 1).
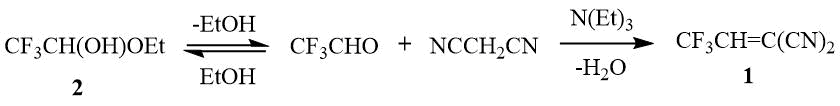
Scheme 1. Synthesis of 2-(2,2,2-trifluoroethylidene)malononitrile 1.
This approach creates the prospect of widespread use of fluorinated cyanoethylenes without their preliminary isolation from the reaction mass. In this paper, we tried to expand a number of used heterocyclic substrates to 4,5,6,7-tetrahydroindole (THI), pyrrole and their N-methyl derivatives. In addition, we also used ethyl ether of trifluoropyruvic acid 3 as a polyfluorocarbonyl precursor of cyanoethylenes. The use of THI instead of indole in reactions with polyfluorocarbonyl compounds makes it possible to ultimately obtain not 3-, but 2-substituted indoles [8]. Unlike indole, THI and pyrrole under mild conditions undergo C2-oxyalkylation under the action of semiacetal 2, which creates competition for the three-component process of alkene 1 formation and C2-alkylation of THI with this alkene.
The interaction of THI or N-Me-THI with aldehyde 2 and malononitrile was carried
out in DCM. In the case of N-Me-THI at 5oC after exposure for 16 hours and subsequent
chromatographic separation, nitrile 4a and alcohol 5a were
obtained with yields of 57% and 17%, respectively. A decrease in the reaction temperature to
-30oC led to the formation of 4A nitrile with a yield of 69% and
a decrease in the yield of 5A alcohol to trace amounts of 4% (scheme 2).
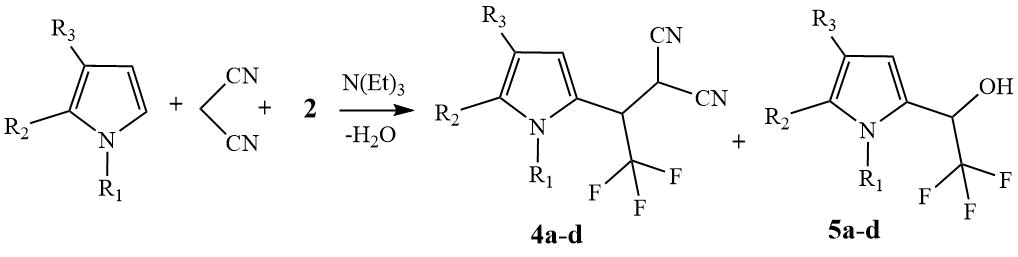
Scheme 2. Three-component synthesis of nitriles 4 and alcohols 5,
a) R1=Me, R2+R3= (CH2)4; b) R1=H, R2+R3= (CH2)4;
c) R1=Me, R2 = R3 = H; d) R1=H, R2 = R3 = H.
At room temperature, the ratio of the resulting products 4a and 5a according to 19F NMR spectroscopy was 43:57, i.e. the formation of alcohol 5a prevailed. From these data, it follows that a decrease in the reaction temperature leads to an increase in the relative amount of nitrile 4a relative to alcohol 5a. The same pattern is observed in the interaction of unsubstituted THI with hemiacetal 2 and malononitrile at 5oC. The relative amounts of isolated products 4b and 5b were 39 and 23%, while at -30oC, only nitrile 4b was obtained with a yield of 75%, and according to 19F NMR spectroscopy and TLC (manifestation of TLC by Ehrlich reagent), no traces of alcohol 5b were found in the reaction mass.
In the case of N-Me-pyrrole, 2 and malononitrile in DCM at 5°C, the three-component reaction leads to the products of C2-alkylation 4c and C2-oxyalkylation 5c, isolated in 54 and 13% yields, respectively. By lowering the reaction temperature to -30°C, the side reaction of C2-oxyalkylation was also completely suppressed, the nitrile yield was 71%. In the case of unsubstituted pyrrole, stable nitrile 4d could not be isolated.
In the case when in the three-component reaction with THI, pyrrole and their N-Me-derivatives,
instead of hemiacetal 2, a more reactive ethyl ester of trifluoropyruvic
acid 3 was used, the products of the three-component reaction, as a rule,
were formed in insignificant amounts (6 - 9%). Relative success was achieved only in the
cases of unsubstituted THI and pyrrole at -30°C. Nitriles 7 and 9 were obtained by this method in 32 and 24% yields, respectively.
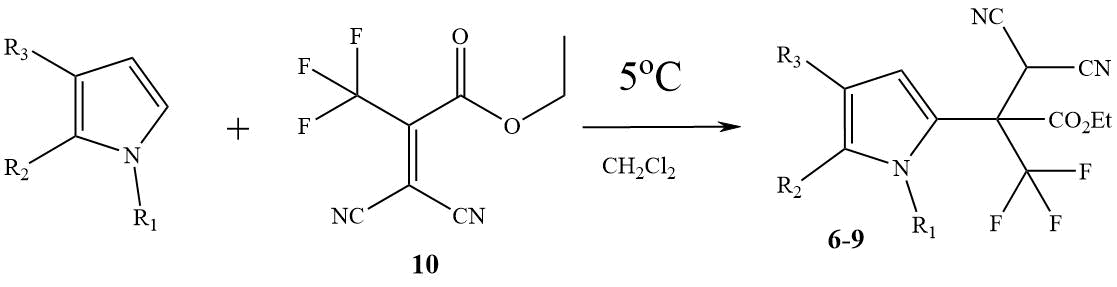
Scheme 3. Synthesis of nitriles 6 - 9 using alkene 10,
6) R1=Me, R2+R3= (CH2)4; 7) R1=H, R2+R3= (CH2)4;
8) R1=Me, R2 = R3 = H; 9) R1=H, R2 = R3 = H.
Unlike synthon 1, alkene 10 is available and has been widely studied earlier using other examples [9]. It turned out that the interaction of THI, pyrrole and their N-Me-derivatives with alkene 10 leads to the products of regioselective C2-alkylation, nitriles 6 - 9, in 77-85% yields (Scheme 3).
Conclusions
Thus, the strategy developed by us for the synthesis of nitriles of hetaryl-2-cyano-4,4,4-trifluorobutanoic acid, based on the use of 2-(2,2,2-trifluoroethylidene)malononitrile, formed in situ from commercially available precursors [4], successfully extended to the synthesis of 2-substituted pyrroles and indoles - nitriles 4a – c and 6 – 9. It has been shown that the presence of hemiacetal 2 in the reaction mass leads to a competing reaction of C2-oxyalkylation of the pyrrole ring, which can be completely suppressed by lowering the reaction temperature to -30°C. In the case of ketoester 3, when interacting with pyrrole, its derivatives and malononitrile, the main reaction is the C2-oxyalkylation of the pyrrole nucleus, rather than its C2-alkylation, formed in situ by alkene 10. Nitriles 6 - 9 were obtained also in high yields from derivatives of pyrrole, THI and alkene 10, previously synthesized from ketoester 3 and malononitrile in 61% yield.
Experimental part
All starting compounds were purchased from Merck, Sigma-Aldrich and used without purification. All eluents and solvents: petroleum ether Tbp = 40-70 oC (PE), EtOAc (EA), DCM and triethylamine were purified by distillation before use. Merck Kieselgel 60 F254 TLC plates were used to monitor the reaction and detect substances. The products were purified by column chromatography using Merck Kieselgel 60 silica gel (0.06-0.20 mm).
1H, 13C JMODECHO and 19F NMR spectra were recorded on a Bruker AvanceTM400 spectrometer at 400, 100, and 376 MHz, respectively. The chemical shifts of protons were determined relative to the residual CDCl3 signals (7.26 ppm) and recalculated to the SiMe4 signal. The 19F NMR spectra were recorded with the suppression of the H-F spin-spin interaction. The chemical shifts of 19F nuclei were determined relative to CFCl3 as an external standard.
Mass spectra were recorded on a Finnigan Polaris Q instrument (ion trap, EI, ionizing electron energy 70 eV) by direct sample injection. High resolution mass spectra were recorded on a Bruker micrOTOF II instrument (electrospray ionization (ESI)). The measurements were performed on positive (capillary voltage 4500 V) or negative (capillary voltage 3200 V) ions. Mass scanning range m/z 50-3000 Da, external or internal calibration (Electrospray Calibrant Solution, Fluka). We used a syringe injection of the substance for solutions in acetonitrile, methanol, or water, the flow rate was 3μL/min. Spray gas nitrogen (4l/min), interface temperature 180οC [10].
General procedure for the preparation of nitriles 4a - d and 6 - 9 by three-component one-stage synthesis
Freshly distilled DCM (2ml), malonitrile (0.1g, 1.5mmol), trifluoro-acetaldehyde ethyl hemiacetal 2 or ethyl trifluoropyruvate 3 (0.216g or 0.255g, respectively, 1.5mmol), triethylamine (0.1g) and 5-7 granules of dried molecular sieves with a pore size of 4A are loaded into a round-bottom flask equipped with a magnetic stir bar. The flask is closed with a septa, vacuumed and filled with argon, cooled to the planned temperature (+5 °C or -30°C) and a pyrrole derivative or THI (1.5mmol) dissolved in 1 ml of DCM is injected with a syringe. The reaction mass is kept at the planned temperature (+5°C or -30°C) overnight. Then the reaction mixture was diluted with a mixture of DCM and PE (10ml, 1/1) and transferred to a chromatographic column containing 100ml of silica gel. The reaction mass is eluted with a mixture of DCM and PE (1/1), gradually increasing the solvent gradient by adding EA to 3-5%. Alcohols 5 are the first to leave the column, their Rf is 0.6-0.9, then nitriles 4 with Rf equal to 0.3-0.6 (PE/EA = 8/2). Solutions of purified products 4 and 5 are evaporated at reduced pressure and re-evaporated with 20 ml of DCM.
General method of synthesis of nitriles 6 - 9 via alkene 10.
Freshly distilled DCM (2ml) and a pyrrole or THI derivative (1.5mmol) are loaded into a round-bottom flask equipped with a magnetic stir bar. The flask is closed with a septa, vacuumed three times consecutively and filled with argon, cooled to +5°C and with using a syringe, alkene 10 (0.327g, 1.5mmol) dissolved in 1ml of DCM is injected. The reaction mass is kept at +5°C for 2-3 hours. Then the reaction mixture was diluted with a mixture of DCM and PE (10ml, 1/1) and transferred on a glass filter containing 10 ml of silica gel. Nitriles 6 - 9 are eluted with a mixture of DCM and PE (1/1), a nitrile solution is collected and evaporated at reduced pressure, then re-evaporated with DCM (20 ml).
(4a) 2-(2,2,2-Trifluoro-1-(1-methyl-4,5,6,7-tetrahydro-1H-indol-2-yl)ethyl)malononitrile
In 3CR procedure at +5оС: yield 57% (0.24g); at -30оС: yield 69% (0.29g). Transparent light yellow slowly solidifying oil, Rf= 0.4 (PE+EА, 0.8+0.2). Spectrum 1Н NMR (CDCl3, δ, ppm., J/Hz): 1.77-1.89 (4H, m, 2CH2); 2.53-2.59 (4H, m, 2CH2); 3.48 (3H, s, Me); 4.21-4.28 (1H, m, CHCF3); 4.38 (1H, d, 6.2 Hz, CH(CN)2); 6.35 (1H, s, 3(С)H THI). Spectrum 13С NMR JMODECHO (CDCl3, δ, ppm., J/Hz): 22.02 (СH2); 22.75 (СH2); 22.95 (СH2); 23.28 (СH2); 25.07 (СH(CN)2); 29.58 (NMe); 41.86 (q, 30.3 Hz, СНCF3); 108.66 (3(С) THI); 110.50 (CN); 110.71 (CN); 116.19; 118.04; 123.69 (q, 281.1 Hz, CF3); 131.20. Spectrum 19F ЯМР (CDCl3, δ, ppm.): -68.31 (CF3). MS (EI, 70 eV), m/z (Irel (%)): 281 [М]+ (2.45); 216 [M - СН(СN)2]+ (100). HRMS (ESI, neg. ions), C14H14F3N3, found m/z: 280.1076 [M - H]-. Calculated: 280.1067.
(4b) 2-(2,2,2-Trifluoro-1-(4,5,6,7-tetrahydro-1H-indol-2-yl)ethyl)malononitrile
In 3CR procedure at +5оС: yield 39% (0.155g); at -30оС: yield 75% (0.3g). Transparent light yellow slowly solidifying oil, Rf= 0.4 (PE+EА, 0.8+0.2). Spectrum 1Н NMR (CDCl3, δ, ppm., J/Hz): 1.74-1.84 (4H, m, 2CH2); 2.48-2.58 (4H, m, 2CH2); 4.00-4.07 (1H, m, CHCF3); 4.34 (1H, d, 4.8 Hz, CH(CN)2); 6.20 (1H, d, 2.4 Hz, 3(C)H THI); 8.08 (1H, br. s., NH). Spectrum 13С ЯМР JMODECHO (CDCl3, δ, ppm., J/Hz): 22.56 (CH2); 22.60 (CH2); 23.05 (CH2); 23.44 (CH2); 25.22 (СH(CN)2); 44.79 (q, 30.3Hz, CHCF3); 110.46 (CN); 110.60 (CN); 111.17 (3(C) THI); 114.15; 118.38; 123.77 (q, 281.4 Hz, CF3); 130.64. Spectrum 19F ЯМР (CDCl3, δ, ppm.): -67.88 (CF3). MS (EI, 70 eV), m/z (Irel (%)): 267 [М]+ (2.1); 202 [M - СН(СN)2]+ (100). HRMS (ESI, neg. ions), C13H12F3N3, found m/z: 266.0921 [M - H]-. Calculated: 266.0911.
(4c) 2-(2,2,2-Trifluoro-1-(1-methyl-1H-pyrrol-2-yl)ethyl)malononitrile
In 3CR procedure at +5оС: yield 54% (0.185g); at -30оС: yield 71% (0.24g). Transparent light yellow slowly solidifying oil, Rf= 0.2 (PE+EА, 0.8+0.2). Spectrum 1Н NMR (CDCl3, δ, ppm., J/Hz): 3.65 (3H, s, NMe); 4.12-4.19 (1H, m, CHCF3); 4.33 (1H, d, 6.6 Hz, CH(CN)2); 6.23 (1H, m, Ar); 6.54 (1H, m, Ar); 6.74 (1H, m, Ar). Spectrum 13С ЯМР JMODECHO (CDCl3, δ, ppm., J/Hz): 25.12 (СH(CN)2); 33.99 (NMe); 41.91 (q, 30.6 Hz, CF3СН); 108.58; 110.16 (CN); 110.46 (CN); 110.75; 118.33; 123.52 (q, 280.7 Hz, CF3); 125.41. Spectrum 19F ЯМР (CDCl3, δ, ppm.): -68.39 (CF3). MS (EI, 70 eV), m/z (Irel (%)): 227 [М]+ (4.9); 162 [M - СН(СN)2]+ (100). HRMS (ESI, neg. ions), C10H8F3N3, found m/z: 226.0591 [M - H]-. Calculated: 226.0598.
(6) Ethyl 2-(dicyanomethyl)-3,3,3-trifluoro-2-(1-methyl-4,5,6,7-tetrahydro-1H-indol-2-yl)propanoate
In 3CR procedure at +5оС: yield 6% (0.03g); at -30оС: yield 9% (0.05g); via alkene 10: yield 81% (0.43g). Transparent light yellow slowly solidifying oil, Rf= 0.3 (PE+EА, 0.8+0.2). Spectrum 1Н NMR (CDCl3, δ, ppm., J/Hz): 1.40 (3H, t, 7.1 Hz, Me); 1.70-1.83 (4H, m, 2CH2); 2.45-2.49 (4H, m, 2CH2); 3.34 (3H, s, NMe); 4.42-4.52 (2H, m, CH2); 4.57 (1H, s, CH(CN)2); 6.17 (1H, s, 3(C)H THI). Spectrum 13С NMR JMODECHO (CDCl3, δ, ppm., J/Hz): 13.78 (CH3); 22.33 (CH2); 22.72 (CH2); 22.95 (CH2); 23.13 (CH2); 29.39 (СH(CN)2); 31.23 (NMe); 64.54 (q, 30.1 Hz, CF3С); 64.94 (CH2); 109.84 (CN); 110.03 (CN); 111.52 (3(C) THI); 115.94; 117.94; 123.20 (q, 280.2 Hz, CF3); 133.26; 164.67. Spectrum 19F NMR (CDCl3, δ, ppm.): -65.30 (CF3). MS (EI, 70 eV), m/z (Irel (%)): 353 [М]+ (3.3); 288 [M - СН(СN)2]+ (100). HRMS (ESI, neg. ions), C17H18F3N3O2, found m/z: 352.1270 [M -H]-. Calculated: 352.1278.
(7) Еthyl 2-(dicyanomethyl)-3,3,3-trifluoro-2-(4,5,6,7-tetrahydro-1H-indol-2-yl)propanoate
In 3CR procedure at +5оС: yield 7% (0.035g); at -30оС: yield 32% (0.165g); via alkene 10: yield 77% (0.39g). Transparent light yellow slowly solidifying oil, Rf= 0.5 (PE+EА, 0.8+0.2). Spectrum 1Н NMR (CDCl3, δ, ppm., J/Hz): 1.43 (3H, t, 7.1 Hz, Me); 1.73-1.81 (4H, m, 2CH2); 2.45-2.56 (4H, m, 2CH2); 4.46-4.56 (2H, m, CH2); 4.76 (1H, s, CH(CN)2); 6.11 (1H, s, 3(C)H THI); 8.45 (1H, br. s., NH). Spectrum 13С NMR JMODECHO (CDCl3, δ, ppm., J/Hz): 13.59 (Me); 22.44 (CH2); 22.50 (CH2); 22.91 (CH2); 23.30 (CH2); 27.46 (СH(CN)2); 58.71 (q, 27.4 Hz, CF3С); 65.15; 109.93; 110.06 (CN); 110.15 (CN); 113.63; 118.38; 122.71 (q, 286.8 Hz, CF3); 131.19; 163.46. Spectrum 19F NMR (CDCl3, δ, ppm.): -67.37 (CF3). MS (EI, 70 eV), m/z (Irel (%)): 339 [М]+ (3.3); 274 [M - СН(СN)2]+ (100). HRMS (ESI, neg. ions), C16H16F3N3O2, found m/z: 338.1121 [M - H]-. Calculated: 338.1122.
(8) Еthyl 2-(dicyanomethyl)-3,3,3-trifluoro-2-(1-methyl-1H-pyrrol-2-yl)propanoate
In 3CR procedure at +5оС: yield 0.0% (0.0g); at -30оС: yield 0.0% (0.0g); via alkene 10: yield 85% (0.38g). Transparent light yellow slowly solidifying oil, Rf = 0.4 (PE+EA, 0.8+0.2). Spectrum 1Н NMR (CDCl3, δ, ppm., J/Hz): 1.38 (3H, t, 7.1 Hz, Me); 3.62 (3H, s, NMe); 4.41-4.50 (2H, m, CH2); 4.70 (1H, s, CH(CN)2); 6.13 (1H, m, Ar); 6.65 (1H, m, Ar); 6.75 (1H, m, Ar). Spectrum 13С NMR JMODECHO (CDCl3, δ, ppm., J/Hz): 13.42 (Me); 29.10 (NMe); 36.23 (СH(CN)2); 58.82 (q, 27.3 Hz, CF3С); 64.17; 106.99; 110.39; 110.40 (CN); 110.61 (CN); 121.93; 123.27; 123.35 (q, 285.2 Hz, CF3); 164.37. Spectrum 19F NMR (CDCl3, δ, ppm.): -66.70 (CF3). MS (EI, 70 eV), m/z (Irel (%)): 299 [М]+ (5.8); 234 [M - СН(СN)2]+ (100). HRMS (ESI, neg. ions), C13H12F3N3O2, found m/z: 298.0801 [M - H]-. Calculated: 298.0809.
(9) Ethyl 2-(dicyanomethyl)-3,3,3-trifluoro-2-(1H-pyrrol-2-yl)propanoate
In 3CR procedure at +5оС: yield 6% (0.025g); at -30оС: yield 24% (0.103g); via alkene 10: yield 79% (0.337g). Transparent light yellow slowly solidifying oil, Rf = 0.4 (PE+EА, 0.8+0.2). Spectrum 1Н NMR (CDCl3, δ, ppm., J/Hz): 1.41 (3H, t, 7.1 Hz, Me); 4.49-4.51 (2H, m, CH2); 4.83 (1H, s, CH(CN)2); 6.27 (1H, m, Ar); 6.41 (1H, m, Ar); 6.90 (1H, m, Ar); 9.07 (1H, br. s., NH). Spectrum 13С NMR JMODECHO (CDCl3, δ, ppm., J/Hz): 13.49 (Me); 27.49 (СH(CN)2); 58.56 (q, 27.6 Hz, CF3С); 65.40; 109.61; 109.95 (CN); 110.03 (CN); 110.81; 116.07; 121.70; 122.59 (q, 286.5 Hz, CF3); 163.26. Spectrum 19F NMR (CDCl3, δ, ppm.): -67.47 (CF3). MS (EI, 70 eV), m/z (Irel (%)): 285 [М]+ (5.4); 220 [M - СН(СN)2]+ (100). HRMS (ESI, neg. ions), C12H10F3N3O2, found m/z: 284.0657 [M - H]-. Calculated: 284.0652.
Acknowledgments
This work was performed with the financial support from Ministry of Science and Higher Education of the Russian Federation. The contribution of Center for molecular composition studies of A.N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences (Moscow, Russia) is gratefully acknowledged. High resolution mass spectrometry was performed in the Department of Structural Studies of N.D. Zelinsky Institute of Organic Chemistry of the Russian Academy of Sciences (Moscow, Russia). For this research trifluoroacetaldehyde ethyl hemiacetal was provided by P&M Invest.
References
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok, H. Liu, The importance of fluorine in the life science industry, Chem. Rev., 2013, 114, 2432–2506. doi: 10.1021/cr4002879.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnely, N. A. Meanwell, Fluorine in Pharmaceuticals: Looking Beyond Intuition, J. Med. Chem., 2015, 58, 8315–8359. doi: 10.1021/acs.jmedchem.5b00258.
- J.-P. Bégué, D. Bonnet-Delpon, Recent advances (1995–2005) in fluorinated pharmaceuticals based on natural products, J. Fluorine Chem., 2006, 127, 992–1012. doi.org/10.1016/j.jfluchem.2006.05.006.
- O. Yu. Fedorovskii, N. D. Chkanikov, Three-component one-step synthesis of new nitriles – 3-(1H-indole-3-yl)-2-cyano-4,4,4-trifluorobutanoic acid, Fluorine Notes, 2021, 134, 3-4. doi: 10.17677/fn20714807.2021.01.02.
- O. Yu. Fedorovskii, A. Yu. Volkonskii, A. S. Golubev, Yu. Ya. Spiridonov, N. D. Chkanikov, Synthesis of ethyl α-nitro-β-trifluoromethyl acrylate and β-trifluoromethyl-substituted tryptophan analogs and their plant growth regulating activity, Russian Chemical Bulletin, 2017, 66, 6, 1116-1121. doi.org/10.1007/s11172-017-1863-z.
- W. J. Middleton, 1,1-Dicyano-2,2-bis(trifluoromethyl)ethylene, J. Org. Chem., 1965, 30, 1402 – 1407. doi.org/10.1021/jo01016a013.
- N. D. Chkanikov and A. S. Golubev, C-Oxyalkylation of Arylamines, Enamines, and Nitrogen-Containing Heterocycles with Polyfluorinated Carbonyl Compounds as a Synthetic Route to Biologically Active Compounds, INEOS OPEN, 2021, 4 (1), 1–19. doi: 10.32931/io2105r.
- Sigan Andrei L., Gusev Dmitrii V., Chkanikov Nikolai D., Shmidt Elena Yu, Ivanov Andrei V., Mihaleva Al’bina I., Hydroxyalkylation of 4,5,6,7-tetrahydroindole with polyfluorocarbonyl compounds as a route to 2-substituted indoles, Tetrahedron Letters, Elsevier BV (Netherlands), 2011, 52, 39, 5025-5028. doi.org/10.1016/j.tetlet.2011.07.071.
- V. Yu. Tyutin, N. D. Chkanikov, A. F. Kolomietz, A. V. Fokin, Synthessis of esters of 3,3-dicyano-2-(trifluoromethyl)acrylic acid and their reactions with arylamines, J. Fluorine Chem., 1991, 51, 323-334. doi.org/10.1016/S0022-1139(00)80188-8.
- Tsedilin A. M., Fakhrutdinov A. N., Eremin D. B., Zalesskiy S. S., Chizhov A. O., Kolotyrkina N. G., Ananikov V. P., How sensitive and accurate are routine NMR and MS measurements?, Mendeleev Comm., 2015, 25, 454. doi.org/10.1016/j.mencom.2015.11.019.
ARTICLE INFO
Received 11 February 2022
Accepted February 2022
Available online February 2022
Recommended for publication by PhD V.L. Don
Fluorine Notes, 2022, 140, 3-4
