Received: June 2021
DOI 10.17677/fn20714807.2021.04.02
Fluorine Notes, 2021, 137, 3-4
REACTIONS OF FLUORINATED 2-(2- OR 4-AMINOARYL)-PROPAN-2-OLS WITH BENZALDEHYDE
T. P. Vasilyeva *, D. V. Vorobyeva
A.N. Nesmeyanov Institute of Organoelement Compounds RAS,
Russian Federation, 119991 Moscow, Vavilov st. 28.
Fax: +7(499) 135 5085. E-mail: d-20@mail.ru
Abstract: The reaction of benzaldehyde with substituted aniline containing a bulky hexafluoroisopropylhydroxy group in p-position in the presence of p-toluenesulfonic acid has synthesized the new bis-CF3-containing benzylidenimines with HO or MeO group. It was shown that substituted aniline with – С(CF3)2OH group in o-position upon reaction with PhCHO under the same conditions undergoes heterocyclization with participation of free NH2- and HO-groups, with formation of a new N-heterocycle-fluoro-substituted dihydrobenzoxazine. The latter exhibits dual reactivity towards alkyl iodides in the presence of cesium carbonate. As a result, N-alkyl-dihydrobenzoxazines and alkoxy-substituted benzylidenimines are formed.
Keywords: hexafluoroacetone, benzaldehyde, C-oxyalkylation of 2,6-xylidines and 2,4-xylidines, benzylidenimines, heterocyclization, fluorine-containing dihydrobenzoxazines.
It is known that organofluorine compounds (especially those containing trifluoromethyl) are now widely used in many domains, including medicine and production of new materials. In this regard, in recent years, the search for effective synthetic methods to introduction of CF3 group into organic compounds has been intensively developing.
Earlier, we reported about use of methyltrifluoropyruvate for introduction of –C(CF3) (OH)- fragment into aliphatic [1, 2], aromatic [1] and heterocyclic [2, 3] compounds. Much attention has recently been paid to sterically hindered anilines with two geminal CF3 groups. Hexafluoroacetone (HFA) is a convenient and available reagent for introducing С(CF3)2OH fragment into arylamines [4-8].
In this paper fluorine-containing methyl-substituted hydroxy-anilines of types (1) and (2) were synthesized, using HFA, and their reactions with benzaldehyde were studied. Anilines 1, 2 are of interest not only as valuable precursors of NHC ligands; the functionalization of these compounds is also relevant in the context of search for bioactive substances, since structures 1 and 2 include the pharmacophore group (CF3)2C(OH)C6H4N. For example, this fragment is contained in some anti-inflammatory drugs (compounds 3 and 4) [9] (see Fig. 1).
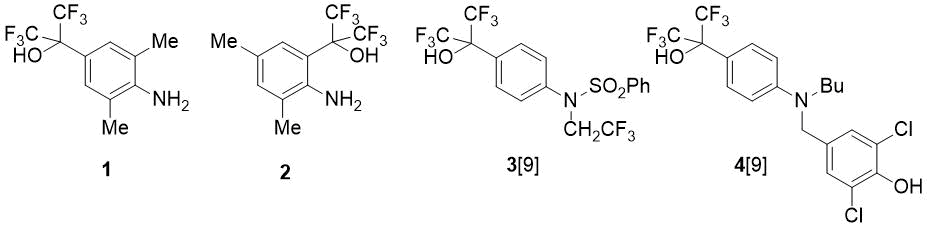
Figure 1. Hexafluoroisopropylhydroxy-substituted anilines.
Fluorine-substituted anilines 1 and 2 were previously obtained by reaction of 2,6- or 2,4-xylidines with gaseous HFA at 170-180°C without solvent [5] or (while synthesis 1) in a closed system in Freon-113 (ClCF2CFCl2) [6], but both of these methods are not high-technological. For synthesis of target anilines 1 and 2, we chose a more convenient method for alkylation of xylidines with commercially available HFA hydrate 1,6H2O (boiling point 104-106°С) in the presence of p-TolSO3H, which was later published in [4]. We have described the improved methods of obtaining 1 and 2 (see Section “Experimental part”).
It is known that for synthesis of fluoroalkyl(aryl)substituted N-heterocyclic carbenes used to create a new catalysts for metathesis, the anilines are required containing alkoxy rather than hydroxy group in fluoroalkyl substituent [4, 7, 8]. In this regard, we studied the reactions of HO-containing anilines 1 and 2 with benzaldehyde to protect the amino function and carry out selective O-alkylation. As it turned out, 2-(4-aminoaryl) propan-2-ol (1) reacts with PhCHO even at room temperature, but complete conversion is not achieved - according to 19F NMR data, a mixture of condensation product (5, 62%) and initial amine (1, 38%). This reaction was completed and pure aldimine 5 was isolated by adding p-toluenesulfonic acid hydrate (see Scheme 1).
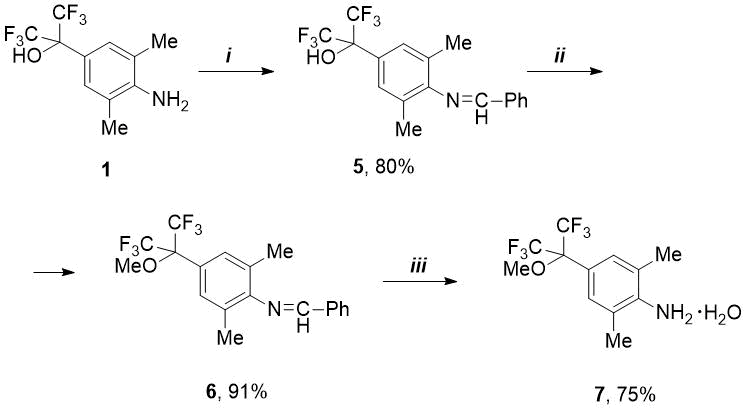
Scheme 1. Reagents and conditions:
i) PhCHO, p-TolSO3H.H2O, 4.5 mol.%, toluene, boiling for 5-7 h;
ii) MeI, K2CO3, MeCN, 55-60 °C, 5h; iii) 4M HCl/Et2O then Na2CO3/H2O.
Then the imine 5 was methylated under mild conditions by means of double excess of methyl iodide in the presence of potassium carbonate. In this way, pure methoxy derivative 6 was obtained in excellent yield without using column chromatography. Compound 6 turned out to be quite stable: the attempts to remove the benzylidene protection under standard conditions by means of dilute HCl (1M or 2M) were unsuccessful. However, under more stringent conditions (4M HCl), it was possible to obtain methoxy-amine hydrochloride, from which the free amine was isolated as a hydrate (compound 7) after treatment with aqueous solution of Na2CO3 (see Scheme 1). A similar anhydrous amine was previously obtained from 2,6-dimethylaniline and HFA in several stages without isolation of intermediates [4].
In order to synthesize the o-alkylated imine, we studied the reaction of PhCHO with isomeric 2-(2-amino-aryl)propan-2-ol (2). The latter, in contrast to amine 1, reacted more vigorously with benzaldehyde: if synthesis 5 required prolonged boiling (5-7 h) in the presence of 4.5 mol% catalyst, then in case of amine 2 the reaction time was only 1 h 10 min when using a smaller amount of p-TolSO3H (1.6 mol.%). However, in this case, instead of expected imine, a new bicyclic fluorine-containing 1,3-benzoxazine (8) was isolated in quantitative yield (see Scheme 2). In literature there are few examples [10-13] of preparation of CF3-substituted benzoxazines (see Fig. 2).
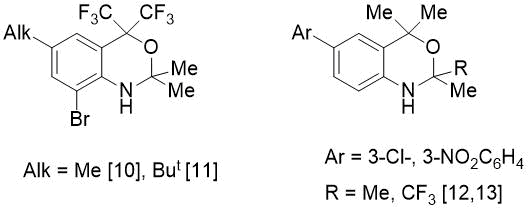
Figure 2. Substituted benzoxazines reported.
Our proposed simple synthesis of NH-oxazine 8 is suitable for scale-up due to availability of starting amino alcohol 2 and the easiness of isolation of 8 in pure form by extraction with hot hexane, without using column chromatography.
In contrast to hydroxy-imine 5, the oxazine 8 synthesized by us did not react with an excess of methyl iodide in the presence of К2CO3 in acetonitrile medium even at elevated temperatures. Under other conditions - with a large excess of MeI (5.2 eq.) and cesium carbonate (3.5 eq.) in DMF medium with prolonged heating (70 °C, 18 h), the conversion of initial 8 was 90%. Herewith, as a result of competing N- and O-methylation, two products were formed – N-methyloxazine 9 and methoxy-imine 10 in a ratio of 2.5:1, which were isolated in low yields using column chromatography (see Scheme 2).
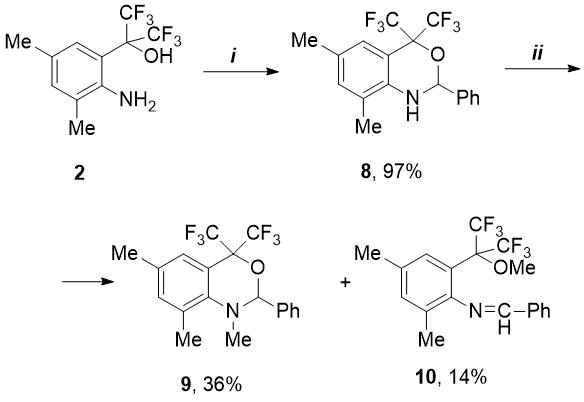
Scheme 2. Reagents and conditions:
i) PhCHO, p-TolSO3H.H2O, 1.6 mol.%, toluene, boiling for 1.2 h;
ii) MeI, Cs2CO3, DMF, 70 °C, 18h.
Further, under the found conditions, we studied the reactions of oxazine 8 with other alkyl halides. We have shown that reaction of 8 with ethyl iodide (3 eq.) in the presence of Cs2CO3 (3 eq.) in DMF medium at 75-80 °C for 20 h leads to 92% conversion of starting compound. In this case, in contrast to methylation, the ratio of obtained ethyl-substituted products, N-ethyloxazine 11 and ethoxy-imine 12, equal to 1:4.3, turned out to be in favor of the latter compound (see Scheme 3). The proposed mechanism for alkylation of oxazine 8 includes the intermediate formation of heterocyclic anion A, which then reacts in two directions. Since methyl halides are more active in SN2 reactions than ethyl halides [14], the predominant N-methylation of anion A occurs with MeI to form N-methyloxazine 9. In the presence of less reactive EtI, the anion A undergoes the rearrangement into alcoholate anion B, stabilized by two CF3 groups , and in this case the main reaction product is ethoxy-imine 12 (see Scheme 3).
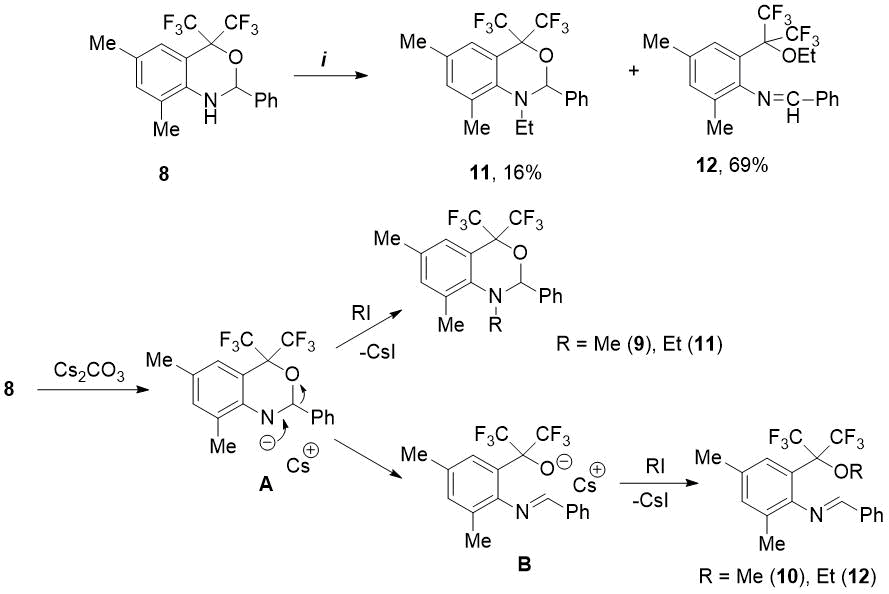
Scheme 3. Reagents and conditions: i) EtI, Cs2CO3, DMF, 75-80°C, 20h.
Attempts to alkylate of oxazine 8 by isopropyl bromide upon heating under various conditions - in the presence of K2CO3 or Cs2CO3 in MeCN, as well as NaH in DMF - were unsuccessful or led to low conversion. Thus, alkyl bromides, unlike iodides, are not active in such alkylation reactions.
Of interest was the preparation of corresponding amine from synthesized ethoxy-imine 12. The latter proved to be more resistant to hydrolysis than p-methoxy-imine 6 (see Scheme 1). Unlike compound 6, o-ethoxy-imine 12 does not react with 4M HCl in diethyl ether. The corresponding ethoxy-amine hydrochloride 13 was prepared by the action of a stronger hydrochloric acid (6M) in ethanol. Free o-ethoxy-amine 14 was prepared from its hydrochloride in quantitative yield by means of excess propylene oxide at room temperature (see Scheme 4).
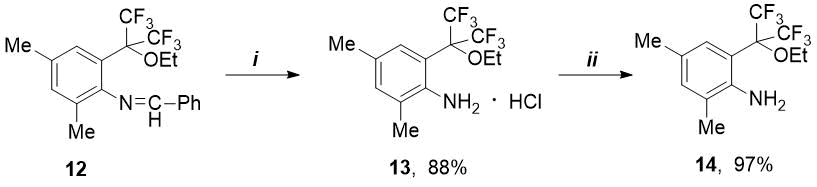
Scheme 4. Reagents and conditions: i) 6M HCl/EtOH, 20C; ii) propylene oxide, 20 °C, 24 h
Hydroxyimine 5, alkoxy-imines 6, 10, 12, benzoxazines 8, 9, 11 and ethoxy-amine 14 synthesized by us are new compounds, the composition and structure of which were proved by the data of elemental analysis and 1H and 19F NMR spectroscopy. In 1H NMR spectra of imines 5, 6, 10 and 12, the signals of benzylidene proton (CH = N) in the range of 7.6-8.2 ppm. are characteristic, while the NH signal of oxazine 8 appears at 4 ppm.
Among compounds obtained, of particular interest are the products of hydrolysis of imines 6 and 12 - sterically hindered arylamines 7 and 14 with bulky hexafluoroisopropylalkoxy groups, which can be used as precursors for synthesis of fluorine-containing N-heterocyclic carbenes and metal complexes, which are, as shown recently [4, 7, 8, 15, 16], the effective catalysts.
Experimental part
NMR spectra were recorded via Bruker Avance-300 and Bruker Avance-400 spectrometers operating at frequencies 300 and 400 MHz (with internal standard SiMe4) for 1H NMR, and 282 and 376 MHz for 19F NMR (CF3COOH), respectively. Elemental analysis was performed at Elemental Analysis Laboratory of INEOS RAS. The reaction progress was monitored by TLC method via Merck plates (silica gel 60 F254, 0.25 mm). Used 2,4- or 2,6-xylidines and HFA hydrate (HFA•1.5H2O) are commercially available reagents. Solvents: petroleum ether (PE), ethyl acetate (EA) and N,N-dimethylformamide (DMF).
2-(4-Amino-3,5-dimethylphenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (1).
To a mixture of 5 g (41.3 mmol) of 2,6-xylidine and 10.37 g (53.7 mmol) of HFA•1.5H2O was added 103.5 mg (0.545 mmol) of p-TolSO3H•H2O, and resulting solution was heated with stirring at 99-103 °С until the reaction is complete (5.5 h). The temperature was brought to room temperature, the solid reaction mixture was treated with ethyl acetate (45 ml) and 1 M NaOH (20 ml) until the solid mixture is dissolve. The organic layer was separated, the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with saturated NaCl solution and dried over MgSO4. The solvent was removed in vacuo, the residue was washed with hexane and dried in vacuo. Yield: 10.56 g (89%) of compound 1 in the form of light lilac powder with melting point (m. p.) 179-180°С (in [5] 174-175 °С, in [6] 173-175°С), RF = 0.46 (solution in Me2CO, in the system Me2CO – СCl4 = 1:3).
1H NMR (300 MHz, CDCl3, , ppm.): 2.27 (s, 6H, 2Me); 2.93 (br. s, 3H, NH2, OH); 7.31 (s, 2H, Ar).
19F NMR (282 MHz, CDCl3, , ppm.): 1.95 (s, 6F, 2CF3).
2-(2-Amino-3,5-dimethylphenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (2).
It was obtained from 2,4-xylidine according to the method described in [4], but instead of 20 h, the heating was carried out for 9 h at 100-105°C. Yield: 81%, in the form of beige crystals. M. p. 113-115°C (in [4] 110-112°C, and in [5] 113-114°C), RF = 0.63 (solution in CHCl3, in EA - PE system = 1:2).
1H NMR (300 MHz, CDCl3, , ppm.): 2.36 (s, 3H, Me); 2.38 (s, 3H, Me); 5.70 (br. s, 3H, NH2, OH); 7.18 (s, 1H, Ar); 7.32 (s, 1H, Ar).
19F NMR (282 MHz, CDCl3, , ppm.): 2.60 (s, 6F, 2CF3).
2-[4-(Benzylideneamino)-3,5-dimethylphenyl]-1,1,1,3,3,3-hexafluoropropan-2-ol (5).
A flask with a Dean-Stark nozzle was charged with 3.0 g (10.45 mmol) of amino alcohol 1, 2.22 g (20.9 mmol) of benzaldehyde, 89 mg (0.47 mmol) of p-TolSO3H•H2O, and 20 ml of toluene. This mixture was reflux with stirring until the water evolution ceased (5-7 h, TLC control in Me2CO – СCl4 system = 1:10). After complete conversion of starting compound, the reaction mixture was evaporated to dryness in a vacuo, the residue was crystallized from hexane and dried in air. Yield: 3.14 g (80%) of compound 5 in the form of white powder. M. p. 135-138°С, RF = 0.48 (solution in CH2Cl2, in system Me2CO – СCl4 = 1:10).
1H NMR (300 MHz, C6D6, , ppm.): 2.06 (s, 6H, 2Me); 3.73 (s, 1H, OH); 7.22-7.24 (m, 3H, Ph); 7.61 (s, 1H, -CH = N); 7.67 (s, 2H, Ar); 7.84-7.87 (m, 2H, Ph).
19F NMR (282 MHz, C6D6, , ppm.): 2.70 (s, 6F, 2CF3).
Founded (%): C, 57.45; H, 3.93; F, 30.56; N, 3.94. C8H15F6NO. Calculated (%): C, 57.60; H, 4.00; F, 30.40; N, 3.73.
N-benzylidene-4-(1,1,1,3,3,3-hexafluoro-2-methoxypropan-2-yl)-2,6 dimethylaniline (6).
To a solution of 2.95 g (7.9 mmol) of hydroxyimine 5 in acetonitrile (20 ml) was added 2.18 g (15.8 mmol) of K2CO3, then 2.24 g (15.8 mmol) of methyl iodide in acetonitrile (8 ml ), and the mixture was heated with vigorous stirring at 55-60°C until the reaction is complete ( 5 h, TLC control in Me2CO – СCl4 system = 1:10). The temperature of this mixture was brought to 20°С, the precipitate was removed by filtration. The mother solution was evaporated in a vacuo, the residue was treated with a mixture of benzene and diethyl ether (20 ml, 1:1). After removal of solid impurities, the filtrate was evaporated to dryness at a pressure of 10 and 1 Torr at 45-70°C. Imine 6 was obtained in the form of heavy brown oil, which solidifies upon cooling to a low-melting light beige mass. Yield: 2.78 g (91%), RF = 0.82 (solution in C6H6 in Me2CO - СCl4 system = 1:10).
1H NMR (300 MHz, C6D6, , ppm.): 2.08 (s, 6H, 2Me); 3.32 (s, 3H, OMe); 7.24-7.26 (m, 3H, Ph); 7.53 (s, 2H, Ar); 7.67 (s, 1H, -CH = N); 7.85-7.88 (m, 2H, Ph).
19F NMR (282 MHz, C6D6, , ppm.): 7.18 (s, 6F, 2CF3).
Founded (%): C, 58.39; H, 4.35; N, 3.78. C19H17F6NO. Calculated (%): C, 58.61; H, 4.37; N, 3.60.
4-(1,1,1,3,3,3-Hexafluoro-2-methoxypropan-2-yl)-2,6 dimethylaniline monohydrate (7).
To a solution of 2.78 g (7.15 mmol) of methoxyimine 6 in diethyl ether (30 ml) was added 12 ml of 4M HCl (48 mmol), and this mixture was vigorously stirred at room temperature until the reaction was complete (with TLC control). The next day, the precipitate that formed was filtered and washed with hexane. Obtained: 1.99 g (83%) of amine hydrochloride in the form of white crystals with m. p. 173-178°С. To isolate the free amine, 16 ml of 15% aqueous solution of Na2CO3 (22.6 mmol) was added to the hydrochloride, and this mixture was stirred at 20°C for 0.5-1 h. The formed precipitate was filtered off, washed with water and hexane and then dried on air. 1.41 g (75% based on hydrochloride) of amine hydrate (compound 7) were obtained in the form of white crystals, m. p. 89-91°С (for anhydrous amine according [4] m. p. is 84-85С) and RF = 0.36 (solution in CDCl3, in system Me2CO – СCl4 = 1:15).
1H NMR (300 MHz, CDCl3, , ppm.): 1.63 (s, 2H, H2O); 2.28 (s, 6H, 2Me); 3.52 (s, 3H, OMe); 3.85 (s, 2H, NH2); 7.17 (s, 2H, Ar).
19F NMR (282 MHz, CDCl3, , ppm.): 6.61 (s, 6F, CF3, 2CF3).
Founded (%): C, 45.47; H, 4.47; F, 35.70; N, 4.36. C12H13F6NOH2O (C12H15F6NO2). Calculated (%): C, 45.14; H, 4.70; F, 35.74; N, 4.39.
6,8-Dimethyl-2-phenyl-4,4-bis(trifluoromethyl)-2,4-dihydro-1H-benzo[d][1,3]oxazine (8).
A mixture of 3 g (10.45 mmol) of amine 2, 1.71 g (16.13 mmol) of benzaldehyde, 32.3 mg (0.17 mmol) of p-TolSO3H•H2O and 15 ml of toluene was boiled with stirring in a flask with Dean-Stark nozzle until the water evolution ceased ( 1 h 10 min) and completion of reaction (TLC control in Me2CO – СCl4 system = 1:6). The brown reaction solution was evaporated to dryness at a pressure of 10 and 1 Torr at 60-80°C. The product was extracted from residue several times with hot hexane. After removal of solvent, 3.81 g (97%) of oxazine 8 were obtained in the form of light cream crystals, m. p. 86-87°С and RF = 0.50-0.54 (solution in CHCl3, in system Me2CO – СCl4 = 1:10).
1H NMR (300 MHz, CDCl3, , ppm.): 2.25 (s, 3H, Me); 2.39 (s, 3H, Me); 4.00 (br. s, 1H, NH); 5.78 (s, 1H, CH); 7.15 (br. s, 1H, Ar); 7.33 (br. s, 1H, Ar); 7.52-7.55 (m, 3H, Ph); 7.68-7.71 (m, 2H, Ph).
19F NMR (282 MHz, CDCl3, , ppm., J/Hz): 1.82 (q, JF-F = 8.8, 3F, CF3); 5.69 (q, JF-F = 8.8, 3F, CF3).
Founded (%): C, 57.68; H, 3.91; N, 3.73. C18H15F6NO.
Calculated (%): C, 57.60; H, 4.00; N, 3.73.
1,6,8-Trimethyl-2-phenyl-4,4-bis(trifluoromethyl)-2,4-dihydro-1H-benzo[d][1,3]oxazine (9).
A mixture of 0.5 g (1.33 mmol) of oxazine 8, 0.98 g (6.9 mmol) of methyl iodide, 1.52 g (4.67 mmol) of cesium carbonate and 4.8 ml of DMF was heated with vigorous stirring at 70°C until the disappearance of bulk original mass 8 ( 18 h, TLC control in PE-EA system = 15:1). The reaction mixture was cooled to 20°С and 12 ml of a mixture of benzene and diethyl ether (1:1) was added. The precipitate was removed by filtration, the mother solution was evaporated at a pressure of 10 and 1 Torr at 45-72°C. The crude product - 0.6 g of brown oil, containing 10% of starting 8, was purified by column chromatography with gradient elution (PE, then PE-EA = 30:1). The dominant product with higher RF was isolated. Yield: 0.19 g (36%) of oxazine 9 in the form of white crystals with m. p. 88-89°C and RF = 0.50 (solution in CHCl3, in PE - EA system = 30:1).
1H NMR (300 MHz, CDCl3, , ppm.): 2.40 (s, 3H, NMe); 2.42 (s, 3H, Me); 2.43 (s, 3H, Me); 5.93 (s, 1H, CH); 7.24 (br. s, 1H, Ar); 7.36 (br. s, 1H, Ar); 7.41-7.56 (m, 3H, Ph); 7.66-7.72 (m, 2H, Ph).
19F NMR (282 MHz, CDCl3, , ppm., J/Hz) 2.87 (q, JF-F = 9.6, 3F, CF3); 5.10 (q, JF-F = 9.6, 3F, CF3).
Founded (%): C, 58.69; H, 4.43; N, 3.56. C19H17F6NO. Calculated (%): C, 58.61; H, 4.37; N, 3.60.
N-benzylidene-2-(1,1,1,3,3,3-hexafluoro-2-methoxypropan-2-yl)-4,6-dimethylaniline (10).
After further elution of this mixture and isolation of oxazine 9 was yielded: 74 mg (14%) of minor product 10 in the form of colorless oil, RF = 0.36 (in PE - EA system = 30:1).
1H NMR (300 MHz, CDCl3 , ppm., J/Hz): 2.44 (s, 6H, 2Me); 2.59 (s, 3H, OMe); 7.21 (br. s, 1H, Ar); 7.28 (br. s, 1H, Ar); 7.40-7.72 (m, 5H, Ph); 8.23 (d, 1H, -CH = N, J = 6.7).
19F NMR (282 MHz, CDCl3 , ppm.): 1.68 (s, 6F, 2CF3).
Founded (%): C, 58.41; H, 4.52; N, 3.78. C19H17F6NO. Calculated (%): C, 58.61; H, 4.37; N, 3.60.
1-Ethyl-6,8-dimethyl-2-phenyl-4,4-bis(trifluoromethyl)-2,4-dihydro-1H-benzo[d][1,3] oxazine (11).
A mixture of 1.51 g (4.03 mmol) of oxazine 8, 1.99 g (12.76 mmol) of ethyl iodide, 3.94 g (12.08 mmol) of cesium carbonate and 12 ml of DMF was heated with vigorous stirring at 75-80 С until the disappearance of bulk original mass 8 ( 20 h, TLC control in PE-EA system = 15:1). After cooling to 20°C, diethyl ether (40 ml) was added to this reaction mixture, and the precipitate was removed by filtration. The mother solution was washed with cold water and dried over MgSO4. After removal of MgSO4, the filtrate was evaporated at a pressure of 10 Torr at 40-71°C. The crude product, 1.64 g in the form of a yellow oil containing 8% of starting 8, was purified by column chromatography with gradient elution (PE-EA = 30:1, 20:1). A minor product with a higher RF was isolated - oxazine 11. Yield: 0.26 g (16%) in the form of white crystals with m. p. 94 – 96 С and RF = 0.68-0.73 (in system PE - EA = 20:1).
1H NMR (400 MHz, CDCl3, , ppm., J/Hz): 0.69 (t, 3H, Me, J = 9.6); 2.43 (s, 6H, 2Me); 2.63-2.76 (m, 1H, CH2); 3.30-3.45 (m, 1H, CH2); 6.00 (s, 1H, CH); 7.24 (s, 1H, Ar); 7.32 (br. s, 1H, Ar); 7.39-7.51 (m, 3H, Ph); 7.61-7.70 (m, 2H, Ph).
19F NMR (376 MHz, CDCl3 , ppm., J/Hz): 2.38 (d, JF-F = 11.3, 3F, CF3); 3.57 (d, JF-F = 11.3, 3F, CF3).
Founded (%): C, 59.54; H, 4.68; N, 3.44. C20H19F6NO. Calculated (%): C, 59.55; H, 4.71; N, 3.47.
N-benzylidene-2-(2-ethoxy-1,1,1,3,3,3-hexafluoropropan-2-yl)-4,6-dimethylaniline (12).
Upon further elution of this mixture (after the isolation of oxazine 11), 1.12 g (69%) of predominant product 12 was obtained in the form of heavy yellow oil, RF = 0.48 (in PE - EA system = 20:1).
1H NMR (400 MHz, CDCl3, , ppm., J/Hz): 1.34 (t, 3H, Me, J = 8.0); 2.15 (s, 3H, Me); 2.41 (s, 3H, Me); 3.79 (q, 2H, OCH2, J = 8.0); 7.24 (s, 1H, Ar); 7.32 (s, 1H, Ar); 7.50-7.65 (m, 3H, Ph); 7.88‑8.02 (m, 2H, Ph); 8.14 (s, 1H, -CH = N).
19F NMR (376 MHz, CDCl3, , ppm.): 7.29 (s, 6F, 2CF3).
Founded (%): C, 59.31; H, 4.56; N, 3.68. C20H19F6NO. Calculated (%): C, 59.55; H, 4.71; N, 3.47.
2-(1,1,1,3,3,3-hexafluoro-2-ethoxypropan-2-yl)-4,6-dimethylaniline (14).
4.9 ml of 6M HCl was added to a solution of 1.19 g (2.95 mmol) of ethoxyimine 12 in 10 ml of ethanol, and this mixture was vigorously stirred at 20С until the reaction is complete (24 h, TLC control in system PE - EA = 15:1). The reaction solution was evaporated to dryness in a vacuo 16 Torr at 50-70 °C, and residue was washed with diethyl ether. Obtained: 0.91 g (2.59 mmol, 88%) of hydrochloride 13 in the form of white powder. To isolate of free amine, the obtained hydrochloride 13 was dissolved in an excess (18 ml) of propylene oxide (14.94 g, 0.26 mol) and stirred at 20°C for 24 h. The reaction mixture was evaporated to dryness at a pressure of 10 Torr at 40 – 73°C. Yield: 0.79 g (97% based on 13) amine 14 in the form of white crystals with m. p. 53-54°С and RF = 0.44 (in system PE - EA = 10:1).
1H NMR (400 MHz, CDCl3, , ppm., J/Hz): 1.42 (t, 3H, Me, J = 8.8); 2.21 (s, 3H, Me); 2.29 (s, 3H, Me); 3.75 (q, 2H, OCH2, J = 8.8); 4.53 (br. s, 2H, NH2); 7.06 (s, 2H, Ar).
19F NMR (376 MHz, CDCl3, , ppm.): 7.24 (s, 6F, 2CF3).
Founded (%): C, 49.29; H, 4.55; N, 4.66. C13H15F6NO. Calculated (%): C, 49.52; H, 4.76; N, 4.44.
Conclusions
Based on the reactions of fluorinated 2-(2- or 4-aminoaryl)propan-2-ols 1 and 2 with benzaldehyde, new fluorine-containing hydroxy- and alkoxy-substituted benzylidenimines 5, 6, 10 and 12 were obtained, which can be used in the synthesis of bis-(CF3)-containing N-heterocycles and amines.
New CF3-containing heterocyclic compounds - NH-, N (Me) - and N (Et) -benzoxazines 8, 9, and 11 - were synthesized by action of benzaldehyde on fluorinated 2-(2-aminoaryl)propan-2-ol 2 followed by alkylation. These compounds are of interest for medicinal chemistry, since the 1,3-oxazine fragment is found both in natural antibiotics [17] and in some synthetic drugs [12, 13].
It was shown that NH-benzoxazine 8 exhibits the dual reactivity with respect to alkyl iodides, giving a mixture of N - (9, 11) and O-alkylation (10, 12) products, the ratio of which depends on the nature of alkyl radical.
Acknowledgments
The work was financially supported by the Ministry of Science and Higher Education of the Russian Federation using the equipment of Center for molecular composition studies of A.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences.
References
- Vasilyeva T.P., Vorobyeva D.V., Russ. Chem. Bull. (Int. Ed.), 2018, 67, 1426.
- Vasilyeva T.P., Vorobyeva D.V., Russ. Chem. Bull. (Int. Ed.), 2020, 69, 300.
- Vorobyeva D.V., Vasilyeva T.P., Osipov S.N., Russ. Chem. Bull. (Int. Ed.), 2018, 67, 1459.
- Masoud S.M., Mailyan A.K., Dorcet V., Roisnel T., Dixneuf P., Bruneau C., Osipov S.N., Organometallics, 2015, 34, 2305.
- Gilbert E., Jones E., Sibilia J., J. Org. Chem., 1965, 30, 1001.
- Chkanikov N.D., Sviridov V.D., Zelenin A.E., Galakhov M.V., Kolomiets A.F., Fokin A.V., Bull. Acad. Sci.USSR, Div.Chem.Sci. (Engl. Transl.), 1990, 39, 323.
- Topchiy M.A., Zotova M.A., Masoud S.M., Mailyan A.K., Ananyev I.V., Nefedov S.E., Asachenko A.F., Osipov S.N., Chem. Eur. J., 2017, 23, 6663.
- Akmalov T.R., Masoud S.M., Petropavlovskikh D.A., Zotova M.A., Nefedov S.E., Osipov S.N., Mendeleev Commun., 2018, 28, 609.
- Chao E., Caravella J., Watson M., Campobasso N., Ghisletti S., Billin A., Galardi C., Wang P., Laffitte B., Iannone M., Goodwin B., Nichols J., Parks D., Stewart E., Wiethe R., Williams S., Smallwood A., Pearce K., Glass C., Willson T., Zuercher W., Collins J., J. Med. Chem., 2008, 51, 5758.
- Nguyen T., Amey R., Martin J., J. Org. Chem, 1982, 47, 1024.
- Nguyen T., Wilson S., Martin J., J. Am. Chem. Soc., 1986, 108, 3803.
- Zhang P., Fensome A., Terefenko E.A., Wrobel J.E., Edwards J.P., Jones T.K., Tegley C.M., Zhi L., Patent US 6358948, 2002.
- Grubb G.S., Zhi L., Jones T.K., Zhang P., Edwards J.P., Fensome A., Terefenko E.A., Wrobel J.E., Tegley C.M., Patent US 6498154, 2002.
- Saunders W., Ionic aliphatic reactions, Prentice-hall, Inc., Englewood cliffs, New York, 1965.
- Masoud S.M., Akmalov T.R., Palagin K.A., Dolgushin F.M., Nefedov S.E., Osipov S.N., Eur. J. Org. Chem., 2018, 5988.
- Masoud S.M., Vorobyeva D.V., Petropavlovskikh D.A., Bruneau C., Osipov S.N., Russ. Chem. Rev., 2021, 90(4), 419.
- Gilchrist T., Heterocyclic chemistry, John Wiley and Sons, Inc., New York, 1992.
ARTICLE INFO
Received 15 June 2021
Accepted 21 June 2021
Available online August 2021
Recommended for publication by PhD M. A. Manaenkova
Fluorine Notes, 2021, 137, 3-4
