Received: December 2020
DOI 10.17677/fn20714807.2021.01.03
Fluorine Notes, 2021, 134, 5-6
An Improved synthesis of 3,3- and 5,5-difluoro-2-aminocyclohexanecarboxylates and extension of the method via organoselenium chemistry
Attila M. Remete,a,b Melinda Nonn,a,b,c Anas Semghouli,a Reijo Sillanpää,d Loránd Kissa,b*
aInstitute of Pharmaceutical Chemistry, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
kiss.lorand@szte.hu
bInterdisciplinary Excellence Centre, Institute of Pharmaceutical Chemistry, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
cMTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, H-6720 Szeged, Eötvös u. 6, Hungary
dDepartment of Chemistry, University of Jyväskylä, FIN-40014, Jyväskylä, Finland
Graphical abstract:
Abstract: A synthetic pathway towards gem-difluorinated cyclohexane β-amino acids, involving regio- and stereoselective hydroxylation, C=C reduction, alcohol oxidation, and deoxofluorination, described earlier has been considerably improved. Across Pd-catalyzed double-bond migration, the intermediate allylic alcohols are directly and efficiently transformed into the corresponding saturated ketones. To extend this improved method, the synthesis of new cyclic β-amino acid derivatives with an allylic alcohol motif was attempted using organoselenium chemistry.
Keywords: β-amino acids, deoxofluorination, double bond migration, oxidative deselenylation, selenocyclization
Introduction
Although fluorine-containing organic compounds are rare in nature [1], they are more and more common in pharmaceutical chemistry [2-5]. The reasons behind the advance of fluorinated drugs are the benefits of fluorination, which originate in the unique properties of the fluorine atom and the carbon–fluorine bond. First of all, because of its small size, the fluorine atom can be used for isosteric replacement of hydrogens, hydroxyl groups or methoxy groups. Importantly, fluorine incorporation can efficiently suppress oxidative metabolism. This is because C–F bonds are stronger than C–H bonds, and the very high electronegativity of fluorine decreases electron density of the surrounding atoms. Fluorine incorporation makes nearby functional groups more acidic, shifting the ratio of charged and neutral drug species. Together with the interesting polar hydrophobic nature of the C–F motif, this shift affects lipophilicity. The fluorine atom of C–F bonds with partial negative charge can also create new attractive electrostatic interactions with target molecules [2,5,6].
From the viewpoint of medicinal chemistry, amino acids are of high importance [7-20]. Within this highly diverse compound family, cyclic β-amino acids have received more and more attention in the last decades [9-13,20]. These compounds can be found as subunits in bioactive natural compounds like blasticidin S (1), amipurimycin (2), gougerotin, and chryscandin (Scheme 1) [9,10,21]. Many small-molecule β-amino acid derivatives, including natural products cispentacin (3) and oxetin as well as synthetic compounds icofungipen and tilidin (4), also possess biological activity (Scheme 1) [9,10]. A rather promising application of cyclic β-amino acids is the construction of β-peptides with high resistance to enzymatic degradation and foldamers [11-13].
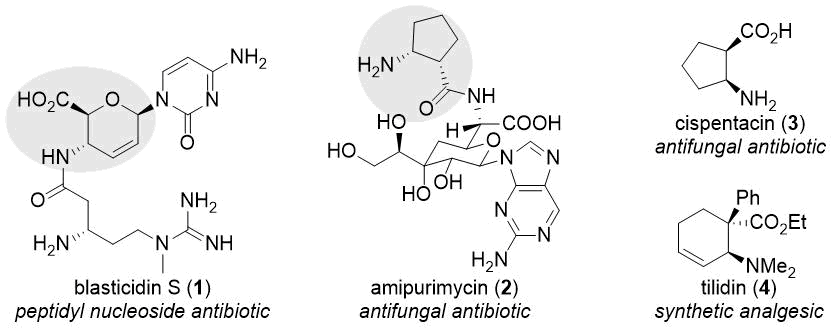
Scheme 1. Bioactive β-amino acid derivatives.
Since both organofluorine compounds and amino acids are of high importance, fluorinated amino acid derivatives have received increasing attention. The most studied compound families are fluorinated acyclic or cyclic α-amino acids and fluorinated acyclic β-amino acids [14-19]. Since fluorinated cyclic β-amino acids are scarcely studied [19-20], we focused on the synthesis of these compounds and developed a considerable number of synthetic pathways towards fluorinated cyclic β-amino acid derivatives in the last decade [22-37].
Results
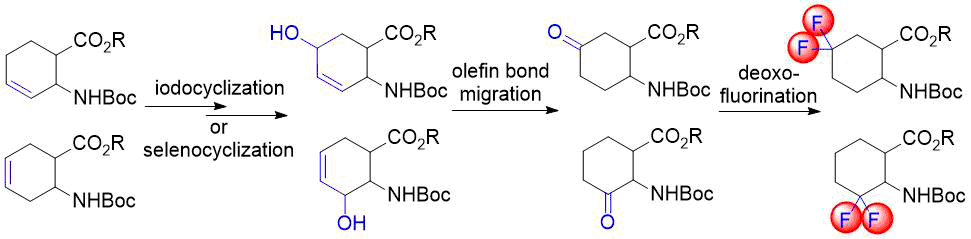
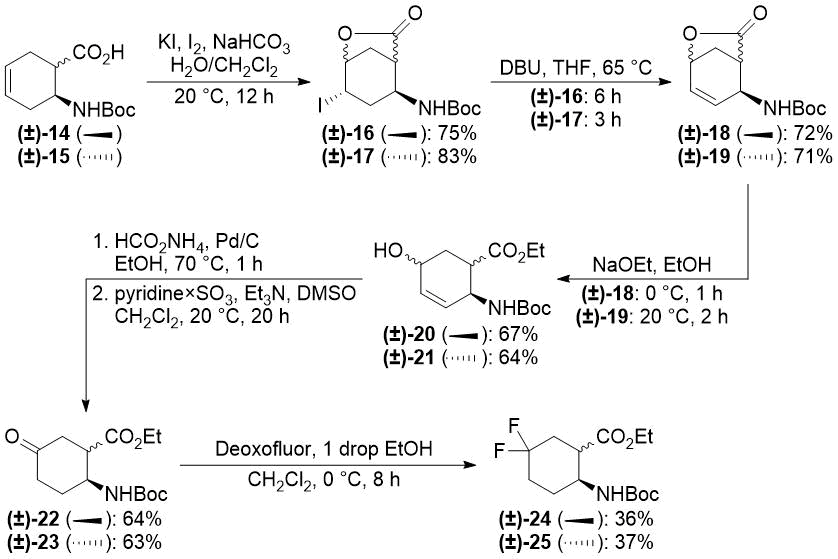
In our earlier reports, we reported the synthesis of gem-difluorinated cyclohexane β-amino acid derivatives from cyclohexene β-amino acids (±)-5 and (±)-14 via regio- and stereoselective hydroxylation, C=C reduction, alcohol oxidation, and deoxofluorination (Schemes 2 and 3) [22,23,38]. The synthesis of gem-difluorinated cyclopentane β-amino acid derivative (±)-37 from cyclopentene β-amino acids (±)-26 and (±)-27 was accomplished in a slightly different pathway (Scheme 4) [26].
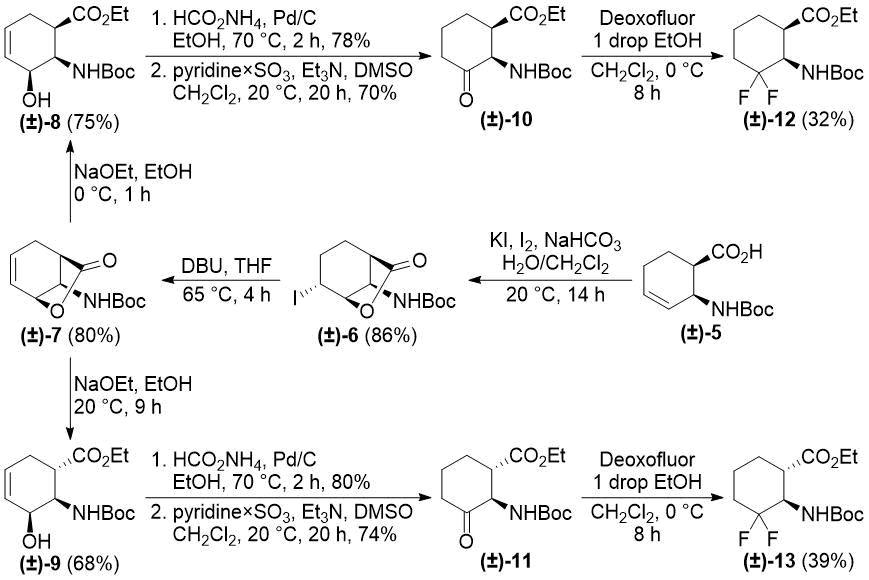
Scheme 2. Original synthesis of 3,3-difluorinated cyclohexane β-amino esters (ref. [22]).
Scheme 3. Original synthesis of 5,5-difluorinated cyclohexane β-amino esters (ref. [23]).
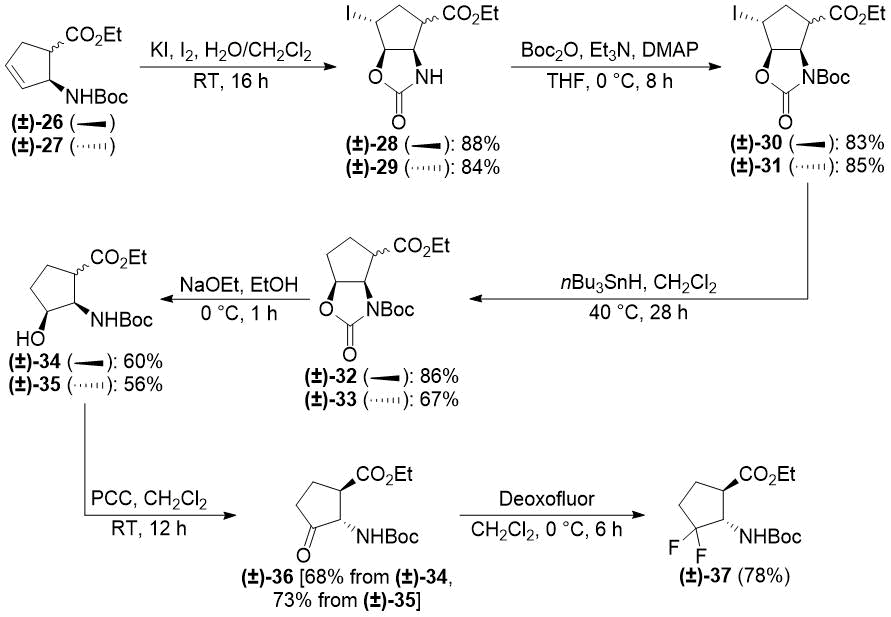
Scheme 4. Original synthesis of gem-difluorinated cyclopentane β-amino ester (±)-37 (ref. [26]).
We realized, however, that these methods have some problematic issues. In the pathways depicted on Schemes 2 and 3, redox-neutral processes (transformations of allylic alcohols (±)-8, (±)‑9, (±)-20, and (±)-21 to ketones (±)-10, (±)-11, (±)-22, and (±)-23) were performed in two subsequent steps. This approach is not completely optimal from an economical viewpoint. Namely, the pathway depicted on Scheme 4 uses stoichiometric amounts of toxic tributyltin hydride and hazardous pyridinium chlorochromate (PCC).
Our aim was to eliminate the above weaknesses as much as possible. First, this directed our attention to transition-metal catalyzed direct transformation of allylic alcohols to ketones. This simple, atom-economic process is based on ring C=C bond isomerization of the substrate to afford an enol intermediate, which tautomerizes to the more stable oxo form (Figure 1). The driving force of the transformation is the higher strength of the C=O bond compared to that of the C=C bond. Such reactions were mostly achieved via homogenous catalysis, but the successful use of heterogeneous catalysts, such as Pd/C [39], Pd/TiO2 [40], Pd/Al2O3 [41-42] or Pd nanoparticles [43], was also reported. Additional benefits are easy handling and separation of heterogeneous catalysts. Therefore, we decided to attempt redox isomerization of allylic alcohols using Pd/C. Possible side reactions are reduction of the substrate C=C bond or the product C=O bond. Consequently, a careful selection of reaction conditions was needed to maximize the yield of the desired product.
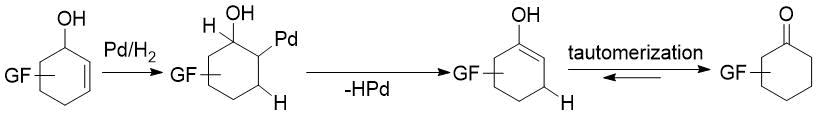
Figure 1. Direct transformation of allylic alcohols to ketones via C=C migration.
Our first model substrate was allylic alcohol derivative (±)-8. After some attempts, some modifications of the conditions, e.g., changing the solvent from EtOH to EtOAc and increasing the reaction time, were sufficient to shift the reaction towards redox isomerization, and product (±)-10 was isolated in 89% yield (Scheme 5). Notably, the original two-step (±)-8 → (±)-10 transformation gave only a 55% overall product yield. The analogous transformation of compound (±)-9 to ketone (±)-11 was also successful (Scheme 5). The isolated yield of 92% compares favorably with 59% of the two-step process. Next, keto esters (±)-10 and (±)-11 were converted. According to a slightly modified procedure, treatment with Deoxofluor in CH2Cl2 at room temperature in 12 h delivered the corresponding difluorinated derivatives (±)-12 and (±)-13 (Scheme 5).
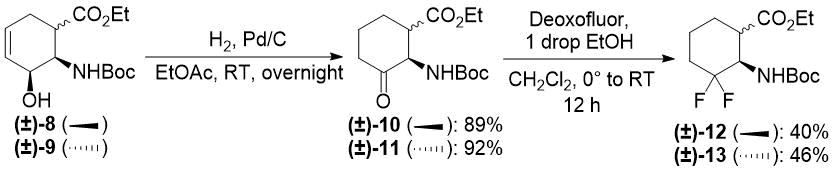
Scheme 5. One-step redox isomerization of allylic alcohols (±)-8 and (±)-9 to the corresponding keto derivatives.
Redox isomerization of allylic alcohols (±)-20 and (±)-21 was also successful (Scheme 6) affording much higher yields than those in the original protocol [(±)-20 → (±)-22: 64% (original two steps) vs. 91% (current protocol); (±)-21 → (±)-23: 63% (original two steps) vs. 90% (new method)]. Both oxo esters (±)-22 and (±)-23 were easily transformed across deoxofluorination into the desired difluorinated products (±)-24 and (±)-25 (Scheme 6).
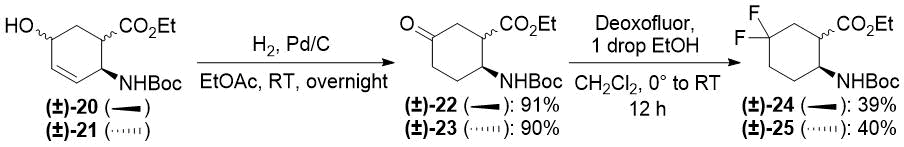
Scheme 6. One-step redox isomerization of allylic alcohols (±)-20 and (±)-21 to keto derivatives.
To extend this new method, we aimed to synthesize other cyclic β-amino esters with allylic alcohol moieties. Unfortunately, subjecting oxazolidinone (±)-30 to HI elimination/heteroring opening (see Schemes 2 and 3) failed, because neither tBuOK nor DBU was able to induce elimination of HI. Treatment of (±)-30 with KOH in THF/H2O provided unexpected product (±)-39 via carbamate ring opening and SN2 substitution of iodide (Scheme 7). Notably, in an earlier study, diol (±)-39 could not be obtained by syn-dihydroxylation of aminoester (±)-26 using OsO4/NMO [44].
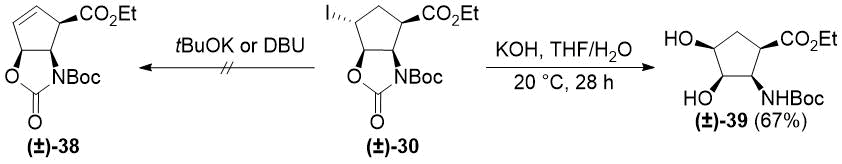
Scheme 7. Reactions of iodooxazolidinone (±)-30 with bases.
In order to solve the elimination problems, we turned to organoselenium chemistry. PhSe+ can take part in similar electrophilic processes as I+, and it can be eliminated under
mild oxidative conditions (e.g., with H2O2) [45-47]. Treatment
of cyclopentene β-amino esters with PhSeBr had an outcome depending on the protecting group.
From N-benzoylated (±)-40, selenooxazoline derivative (±)-41 was formed, while carbamate protecting groups (especially Boc) led to the formation of selenooxazolidinone
(±)-44. N-Boc protection of compound (±)-44 followed
by oxidative deselenylation under basic conditions [47] yielded the desired product (±)-38 (Scheme 8).
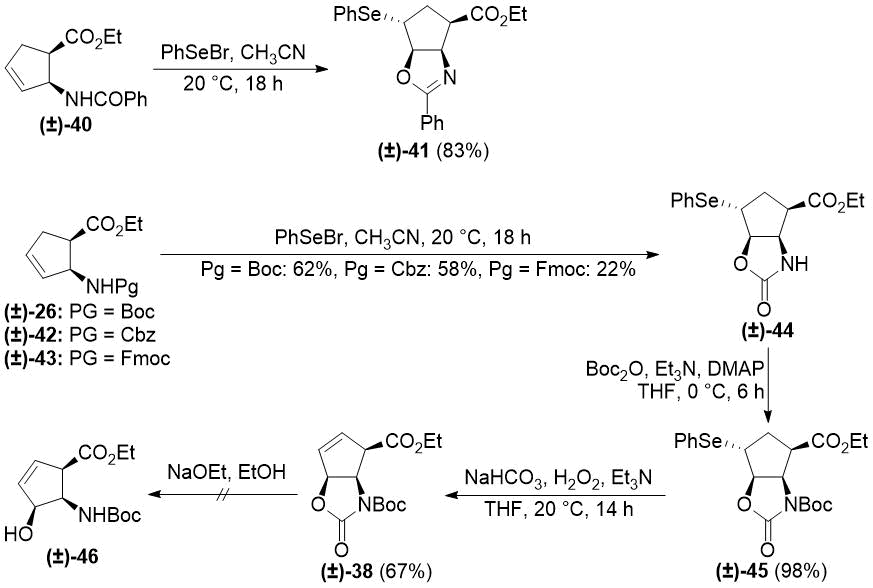
Scheme 8. Selenocyclizations of cis cyclopentene β-amino esters.
Unfortunately, all attempts to open the heteroring of (±)-38 with ethoxide resulted in decomposition with the formation of unidentifiable polymeric materials.
Analogous transformations of N-Boc-protected trans aminoester (±)-27 afforded unsaturated oxazolidinone (±)-49 (Scheme 9). In addition to NMR analysis, the structure of intermediate (±)-48 was confirmed by its X-ray crystal structure (Figure 2). Details of its structure determination are presented in Experimental Section. Similar to its stereoisomer (±)-38, subjecting (±)-49 to ethoxide treatment resulted in decomposition instead of heteroring opening.
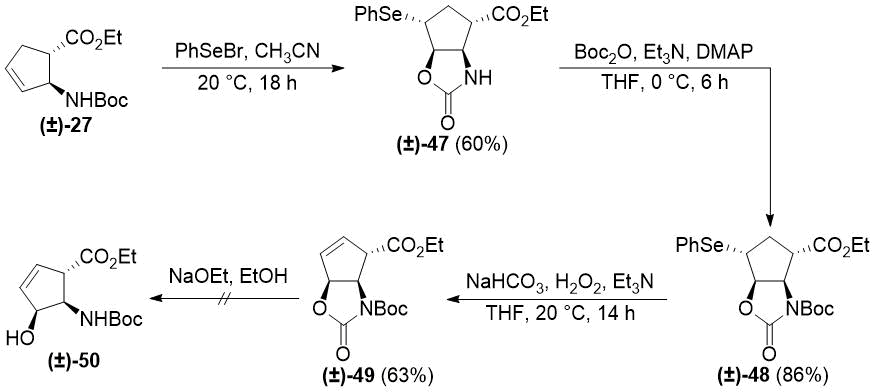
Scheme 9. Selenocyclization of trans cyclopentene β-amino ester (±)-27.
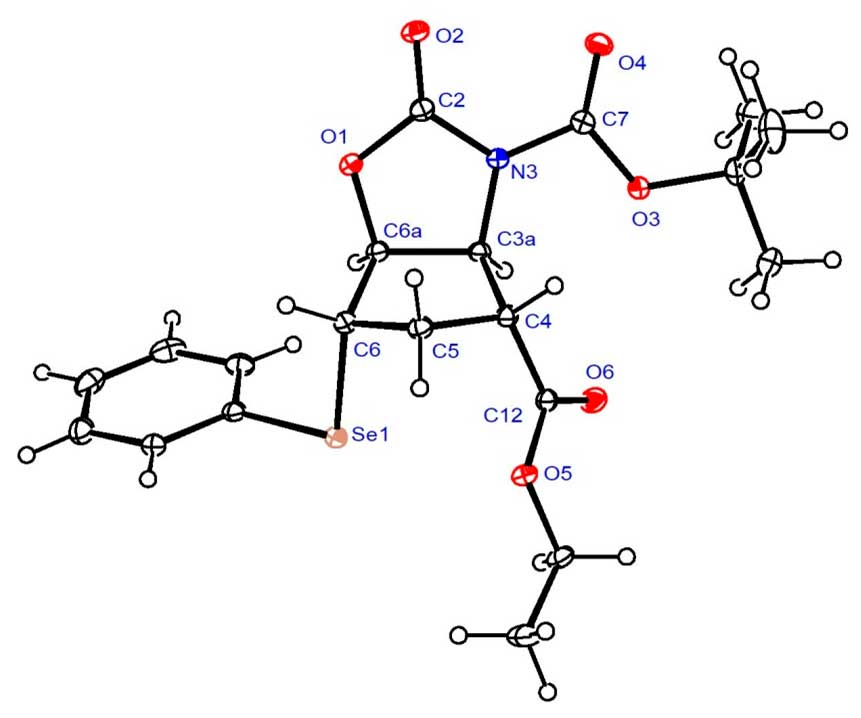
Figure 2. Crystal structure of selenooxazolidinone (±)-48.
Next, we applied the selenocyclization/N-protection/oxidative deselenylation sequence to cyclohexene β-amino ester (±)-51. Unexpectedly, the oxidation led to the formation of highly unsaturated ester (±)-55. The most plausible explanation is a base-promoted E1cb elimination of the desired product (±)-54 (Scheme 10). Decomposition of (±)‑38 and (±)-49 upon treatment with the strongly basic ethoxide can be explained with similar E1cb elimination processes (the resulting highly reactive α,β,γ,δ-unsaturated esters easily undergo polymerization).
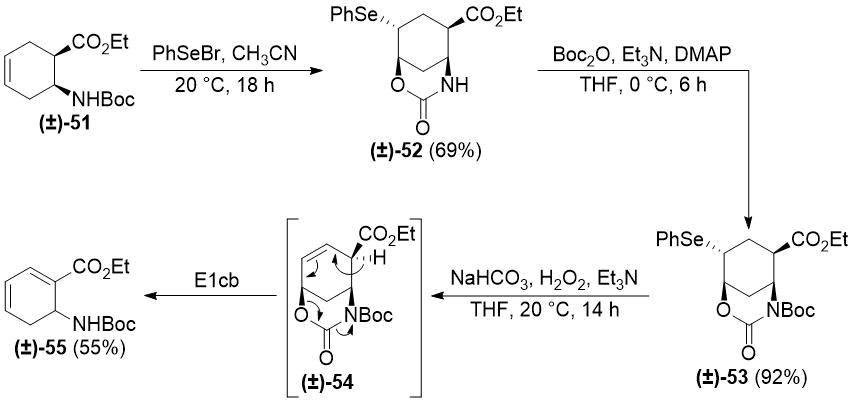
Scheme 10. Selenocyclization of cyclohexene β-amino ester (±)-51.
To avoid E1cb elimination, a possible solution is to perform the heteroring opening before the oxidative deselenylation and replacing the poor leaving group hydroxide with the acceptable leaving group carbamate. Even in this case, it may be necessary to perform the oxidation under acidic conditions (e.g., MCPBA or H2O2/CF3COOH [46]) instead of the basic conditions applied above. These studies are currently in progress in our laboratory.
Conclusions
Using one-step redox-neutral isomerization of allylic alcohols into saturated ketones, we significantly improved the synthesis of the cis and trans isomers of 3,3- and 5,5-difluorinated 2-aminocyclohexanecarboxylates. To expand this improved strategy, preparation of various β-aminocycloalkanecarboxylates with an allylic alcohol motif was attempted using organoselenium chemistry. Some key intermediates were prone to E1cb elimination and the desired allylic alcohol products were not formed. Modification of the synthetic pathway to eliminate the source of the problem is still ongoing. Further variations of the synthetic methods in view of the extension of the methods to access geminal difluorinated scaffolds are currently under investigation.
Experimental
General information
Chemicals were purchased from Sigma-Aldrich. Solvents were used as received from the suppliers. Melting points were determined with a Kofler apparatus. Elemental analyses were performed with a Perkin–Elmer CHNS-2400 Ser II elemental analyser. Silica gel 60 F254 was purchased from Merck. NMR spectra were acquired at room temperature on a Bruker Avance 400 spectrometer (1H frequency 400.13 MHz; 13C frequency 100.76 MHz) in CDCl3 or D6-DMSO solution, using the deuterium signal of the solvent to lock the field. The 1H and 13C chemical shifts are given relative to TMS. β-Amino acid derivatives (±)-5-(±)-37, (±)-40, (±)-42, (±)-43 and (±)-51 are known compounds earlier described in the literature [22,23,26,38,48-50].
General procedure for Pd-catalyzed isomerization
To a solution of 1 mmol allylic alcohol in 10 ml EtOAc 15 mg Pd/C (10 wt %) was added. The reaction mixture was stirred at 20 °C under hydrogen (1 bar) overnight. The reaction mixture was then filtered on a fritted glass filter covered with Celite®. The filtrate was dried (Na2SO4) and concentrated. The crude product was purified by column chromatography on silica gel (n‑hexane/EtOAc).
General procedure for selenocyclizations
To a solution of 5 mmol β-amino ester in 25 ml MeCN 5 mmol PhSeBr was added and the mixture was stirred at 20°C for 18 h. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was diluted with 60 ml EtOAc and washed with 2×20 ml saturated aqueous NaHCO3 solution. The organic phase was dried (Na2SO4) and concentrated. The crude product was crystallized from Et2O.
General procedure for N-Boc protection of selenocyclization products
2 Equiv Et3N, 1 equiv Boc2O, and 20 mol% DMAP were added to a solution of 1.5 g selenocyclized compound in 30 ml THF at 0°C. The resulting mixture was allowed to warm up to 20°C and stirred for 6 h. Then, the reaction mixture was diluted with 70 ml EtOAc and washed with 3×30 ml water. The organic phase was dried (Na2SO4) and concentrated. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc).
General procedure for oxidative deselenylation.
To a solution of 2 mmol phenylselenyl β-amino ester in 25 ml THF, 2 equiv NaHCO3, and 5 equiv H2O2 (as 30% aqueous solution) were added at 0°C. The mixture was allowed to warm up to 20°C and stirred for 30 min. After adding 2.5 equiv Et3N stirring was continued for 14 h. Then the reaction mixture was diluted with 90 mL EtOAc and washed with 2×20 ml water. The organic phase was dried (Na2SO4) and concentrated. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc).
Reaction of iodooxazolidinone (±)-30 with KOH/THF.
To a solution of 300 mg compound (±)-30 in 15 ml THF, 1 ml water, and 2 equiv KOH were added and the mixture was stirred at 20°C for 28 h. Then the reaction mixture was diluted with 20 ml water and extracted with 3×10 ml CH2Cl2. The organic phase was dried (Na2SO4) and concentrated. The crude product was purified by column chromatography on silica gel (n‑hexane/EtOAc).
General procedure for fluorination of keto-amino esters.
To a solution of amino ester (±)-10, (±)-11, (±)-22 or (±)-23 (1.5 mmol) in CH2Cl2 (15 ml) one drop of EtOH was added followed by the dropwise addition of Deoxofluor (1.5 equiv, 50% in toluene) at 0°C and stirring was continued at room temperature overnight. The mixture was then diluted with CH2Cl2 (25 ml) and the organic phase was washed with an aqueous solution of NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the crude product was purified by column chromatography on silica gel (n-hexane-EtOAc 4:1).
X-ray crystallographic study of (±)-48.
Crystallographic data were collected at 123 K with a Nonius-Kappa CCD area detector diffractometer, using graphite-monochromatized Mo-K radiation ( = 0.71073 Å) as reported earlier [51]. The structure was solved by direct methods by use of the SHELXS-97 program [2] and full-matrix, least-squares refinements on F2 were performed by use of the SHELXL-97 program [52]. The CH hydrogen atoms were included at fixed distances from their host atoms with the fixed displacement parameters. The ORTEP plot was drawn with ORTEP-3 for Windows [53]. The deposition number CCDC 2051318 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: (internat.) + 44-1223-336-033; E-mail: deposit@ccdc.cam.ac.uk)
Crystal data for (±)-48, C20H25NO6Se, Mr = 454.37, monoclinic, space group P21/c (no. 14), a = 9.1060(1), b = 18.8854(3), c = 11.8729(2) Å, = 90, β = 90.73(1), = 90, V = 2041.62(5) Å3, T = 123 K, Z = 4, μ(Mo-Kα) = 1.478 mm-1. Total 4450 refelections, unique 3973. Refinement of 4450 reflections (257 parameters) with I > 2(I) converged at final R1 = 0.0236 ( R1 all data = 0.0289), wR2 = 0.0553 (wR2 all data = 0.0575), GOF = 1.049.
See PDF version for supporting Information with NMR spectra.
Acknowledgments
We are grateful to the Hungarian Research Foundation (NKFIH No K 119282) for financial support. The financial support of the GINOP-2.3.2-15-2016-00038 project is also acknowledged. This research was supported by the EU-funded Hungarian grant EFOP-3.6.1-16-2016-00008. Ministry of Human Capacities, Hungary (grant 20391-3/2018/FEKUSTRAT) is also acknowledged.
References
- Carvalho, M. F., Oliveira, R. S., Crit. Rev. Biotechnol., 2017, 37, 880-897.
- Hagmann, W. K. J., Med. Chem., 2008, 51, 4359-4369.
- Wang, J., Sánchez-Roselló, M., Aceña, J. L., del Pozo, C., Sorochinsky, A. E., Fustero, S., Soloshonok, V. A., Liu, H., Chem Rev., 2014, 114, 2432-2506.
- Zhou, Y., Wang, J., Gu, Z., Wang, S., Zhu, W., Aceña, J. L., Soloshonok, V. A., Izawa, K., Liu, H., Chem Rev., 2016, 116, 422-518.
- Han, J., Remete, A. M., Dobson, L. S., Kiss, L., Izawa, K., Moriwaki, H., Soloshonok, V. A., O’Hagan, D. J., Fluorine Chem., 2020, 239, 109639.
- Purser, S., Moore, P. R., Swallow, S., Gouverneur, V., Chem. Soc. Rev., 2008, 37, 320-330.
- Soloshonok, V. A., Izawa, K. (Eds.), Asymmetric Synthesis and Application of α-Amino Acids, ACS Symposium Series #1009, Oxford University Press, 2009.
- Liu, J., Han, J., Izawa, K., Sato, T., White, S., Meanwell, N. A., Soloshonok, V. A., Eur. J. Med. Chem., 2020, 208, 112736.
- Kiss, L., Fülöp, F. Chem Rev. 2014, 114, 1116-1169.
- Kiss, L., Mándity, I. M., Fülöp, F., Amino Acids, 2017, 49, 1441-1455.
- Martinek, T. A., Fülöp, F., Chem. Soc. Rev., 2012, 41, 687-702.
- Gopalan, R. D., Del Borgo, M. P., Mechler, A. I., Perlmutter, P., Aguilar, M.-I., Chem. Biol., 2015, 22, 1417-1423.
- Lee, M.-R., Raman, N., Gellman, S. H., Lynn, D. M., Palecek, S. P., ACS Chem. Biol., 2014, 9, 1613-1621.
- Salwiczek, M., Nyakatura, E. K., Gerling, U. I. M., Ye, S., Koksch, B., Chem. Soc. Rev., 2012, 41, 2135-2171.
- Moschner, J., Stulberg, V., Fernandes, R., Huhmann, S., Leppkes, J., Koksch, B., Chem. Rev., 2019, 119, 10718-10801.
- Qiu, X.-L., Qing, F.-L., Eur. J. Org. Chem., 2011, 3261-3278.
- Mei, H., Han, J., Klika, K. D., Izawa, K., Sato, T., Meanwell, N. A., Soloshonok, V. A., Eur. J. Med. Chem., 2020, 186, 111826.
- March, T. L., Johnston, M. R., Duggan, P. J., Gardiner, J., Chem. Biodivers., 2012, 9, 2410‑2441.
- Mikami, K., Fustero, S., Sánchez-Roselló, M., Aceña, J. L., Soloshonok, V., Sorochinsky, A., Synthesis, 2011, 19, 3045-3079.
- Kiss, L., Fülöp, F., Chem. Rec., 2018, 18, 266-281.
- Wang, S., Zhang, Q., Zhao, Y., Sun, J., Kang, W., Wang, F., Pan, H., Tang, G., Yu, B., Angew. Chem. Int. Ed., 2019, 58, 10558-10562.
- Kiss, L., Forró, E., Fustero, S., Fülöp, F., Org. Biomol. Chem., 2011, 9, 6528-6534.
- Kiss, L., Forró, E., Fustero, S., Fülöp, F., Eur. J. Org. Chem., 2011, 4993-5001.
- Nonn, M., Kiss, L., Hänninen, M. M., Sillanpää, R., Fülöp, F., Chem. Biodivers., 2012, 9, 2571-2580.
- Kiss, L., Nonn, M., Sillanpää, R., Fustero, S., Fülöp, F. Beilstein J., Org. Chem., 2013, 9, 1164‑1169.
- Kiss, L., Nonn, M., Forró, E., Sillanpää, R., Fustero, S., Fülöp, F., Eur. J. Org. Chem., 2014, 19, 4070-4076.
- Nonn, M., Kiss, L., Haukka, M., Fustero, S., Fülöp, F., Org. Lett., 2015, 17, 1074-1077.
- Ábrahámi, R. A., Kiss, L., Barrio, P., Fülöp, F., Tetrahedron, 2016, 72, 7526-7535.
- Kiss, L., Remete, A. M., Nonn, M., Fustero, S., Sillanpää, R., Fülöp, F., Tetrahedron, 2016, 72, 781-787.
- Kiss, L., Nonn, M., Sillanpää, R., Haukka, M., Fustero, S., Fülöp, F., Chem. Asian J., 2016, 11, 3376-3381.
- Remete, A. M., Nonn, M., Fustero, S., Fülöp, F., Kiss, L., Molecules, 2016, 21, 1493.
- Kiss, L., Petrovszki, Á., Vass, C., Nonn, M., Sillanpää, R., Haukka, M., Fustero, S., Fülöp, F., Chemistry Select, 2017, 2, 3049-3052.
- Remete, A. M., Nonn, M., Fustero, S., Haukka, M., Fülöp, F., Kiss, L., Beilstein J. Org. Chem., 2017, 13, 2364-2371.
- Remete, A. M., Nonn, M., Fustero, S., Haukka, M., Fülöp, F., Kiss, L., Eur. J. Org. Chem., 2018, 27-28, 3735-3742.
- Nonn, M., Fülöp, F., Kiss, L., Fluorine Notes, 2018, 1(116), 1-2, DOI: 10.17677/fn20714807.2018.01.01
- Ouchakour, L., Nonn, M., Kiss, L. Fluorine Notes 2019, 1(122), 1-2, DOI: 10.17677/fn20714807.2019.01.01
- Remete, A. M., Benke, Z., Kiss, L. Fluorine Notes 2019, 6(127), 3-4, DOI: 10.17677/fn20714807.2019.06.02
- Forró, E., Schönstein, L., Kiss, L., Vega-Peñaloza, A., Juaristi, E., Fülöp, F., Molecules, 2010, 15, 3998-4010.
- Musolino, M. G., Cutrupi, C. M. S., Donato, A., Pietropaolo, D., Pietropaolo, R., Appl. Catal. A, 2003, 243, 333-346.
- Musolino, M. G., De Maio, P., Donato, A., Pietropaolo, R., J. Mol. Catal. A: Chem., 2004, 208, 219-224.
- Zsolnai, D., Mayer, P., Szőri, K., London, G., Catal. Sci. Technol., 2016, 6, 3814-3820.
- Dékány, A., Lázár, E., Szabó, B., Havasi, V., Halasi, G.,· Sápi, A., Kukovecz, Á., Kónya, Z.,·Szőri, K., London, G. Catal. Lett., 2017, 147, 1834-1843.
- Sadeghmoghaddam, E., Gu, H., Shon, Y.-S., ACS Catal., 2012, 2, 1838-1845.
- Benedek, G., Palkó, M., Wéber, E., Martinek, T. A., Forró, E., Fülöp, F., Eur. J. Org. Chem., 2008, 21, 3724-3730.
- Wirth, T. (Ed.), Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, Germany, 2012.
- Remete, A. M., Novák, T. T., Nonn, M., Haukka, M., Fülöp, F., Kiss, L., Beilstein J. Org. Chem., 2020, 16, 2562-2575.
- Parsons, P. J., Camp, N. P., Edwards, N., Sumoreeah, L. R., Tetrahedron, 2000, 56, 309-315.
- Kiss, L., Forró, E., Sillanpää, R., Fülöp, F., J. Org. Chem., 2007, 72, 8786-8790.
- Coldham, I., Price, K. N., Rathmell, R. E., Org. Biomol. Chem., 2003, 1, 2111-2119.
- Fülöp, F., Palkó, M., Forró, E., Dervarics, M., Martinek, T. A., Sillanpää, R., Eur. J. Org. Chem., 2005, 3214-3220.
- Kanizsai, I., Szakonyi, Z., Sillanpää, R., D'Hooghe, M., De Kimpe, N., Fülöp, F., Tetrahedron: Asymmetry, 2006, 17, 2857.
- Sheldrick, G.M., A short history of SHELX., Acta Cryst., 2008, A64, 112-122.
- Farrugia, L. J. J., Appl. Cryst., 1997, 30, 565.
ARTICLE INFO
Received 21 December 2020
Accepted 12 January 2021
Available online February 2021
Recommended for publication by Prof. S.M. Igumnov
Fluorine Notes, 2021, 134, 5-6
