Received: August 2020
DOI 10.17677/fn20714807.2020.05.03
Fluorine Notes, 2020, 132, 5-6
THEORETICAL ASSESSMENT OF ACID STRENGTH OF ANTIVIRAL FAVIPIRAVIR MEDICATION
V.A. Babkin1, D.S. Andreev1, Yu.A. Vashuta1, A.V. Kozhukhova1, V.S. Belousova2, E.S. Titova3,4, A.R. Titova4, A.I. Rakhimov3, A.K. Brel4, R.O. Boldyrev5, M.I. Artsis5, G.E. Zaikov5
1Volgograd State Technical University (Sebryakovsky br.),403343 Volgograd Region, Mikhailovka, Michurina st., 21.
e-mail: babkin_v.a@mail.ru
2I.M. Sechenov 1st Moscow State Medical University, 119991, st. Trubetskaya, 8, bld. 2.
e-mail: desdemosha@mail.ru
3Volgograd State Technical University,400005 Volgograd, Lenin av., 28.
e-mail: organic@vstu.ru
4Volgograd State Medical University,400131 Pavshikh Bortsov sq., 1.
e-mail titova051@rambler.ru
5N. M. Emanuel Institute of Biochemical Physics RAS,119334, Moscow, Kosygina st., 4.
e-mail: chembio@sky.chph.ras.ru
Abstract: AB INITIO and DFT methods were used to perform a quantum-chemical calculation of antiviral drug favipiravir and theoretically estimated its acidic strength (pKa = 13-14, where pKa is a universal indicator of acidity). It was found that this antiviral medication belongs to the class of weak acids (9 <pKa <14). It has been suggested that a decrease in acidic strength of favipiravir may increase its effectiveness.
Keywords: favipiravir, acid strength, quantum-chemical calculation, AB INITIO method, DFT method, pKa.
Introduction
Favipiravir (6-fluoro-3-hydroxypyrazine-2-carboxamide, C5H4FN3O2) is an antiviral medication (drug) developed in Japan for treatment of viral diseases, including influenza viruses, rhinovirus, respiratory syncytial virus and other life-threatening viral diseases [1]. The principle of its action is based on elongation inhibition of forming RNA chain [2]. Currently, favipiravir is being studied as a potential medication for treatment other viral infections, including coronavirus infection (COVID-19) [3]. Therefore, the question of increasing the effectiveness of this medication currently remains open. One of the ways to increase the effectiveness of favipiravir (and any other medical drug), as it appears to the authors, is the ability to control the acidic strength of this medication in accordance with well-known rule: when the acidic strength of any chemical compound decreases, its selectivity increases (to a certain limit), while maintaining its sufficiently high activity [4].
In this regard, the aim of this work is to theoretically estimate the acidic strength of studied compound - 6-fluoro-3-hydroxypyrazine-2-carboxamide - by quantum-chemical calculation of its optimal geometric and electronic structure and obtaining various quantum-chemical parameters (the total energy of molecular system C5H4FN3O2 (Eо), electron energy (Eel), distribution of atoms electron density (qA), where qmaxH+ is the maximum hydrogen atom charge), etc.. These parameters can be used in other studies, for example, when studying the mechanisms of chemical and biochemical reactions in which favipiravir can participate.
Methodical part
Two methods - AB INITIO/6-311G* and DFT-PBEO/6-311G** - for quantum-chemical calculation of studied drug favipiravir were chosen. The calculation was carried out according to programs [5-7], similar to [8]. The choice of these methods was due to the fact that at present it reproduce both the energy characteristics of favipiravir and the distribution of its atoms electron density quite well and with high accuracy. The calculations were carried out within framework of gas phase molecular model [9-10].
Calculation data
The optimized geometric and electronic structure of favipiravir's molecule is shown in Figs. 1, 2. The quantum-chemical parameters (E0, Eel, qmaxH+) and pKa of studied favipiravir are presented in Table 1. For model of favipiravir obtained by AB INITIO method, the optimized bond lengths between carbon atoms in the ring are within the range 1.38-1.51 Å, С-О-1.19-1.32 Å,С-Н-1.07 Å, C-N-1.29-1.35 Å, F-C-1.32 Å, H-N-0.99 Å, H-O-0.94 Å.
The following corresponding optimized bond angles (in degrees) were obtained: N(6)-C(2)-C(1)-119,C(5)-N(6)-C(2)-119, C(2)-C(1)-N(3)-121, C(1)-N(3)-C(4)- 119,N(3)-C(4)-C(5)-120, C(4)-C(5)-N(6)-122, C(4)-C(5)-F(7)-120, C(2)-C(1)-O(8)- 122, N(6)-C(2)-C(9)-117, C(1)-C(2)-C(9)-124, C(2)-C(9)-N(10)-114, O(11)-C(9)-N(10)-124, C(2)-C(9)-O(11)-122, N(3)-C(4)-H(12)-119, C(1)-O(8)-H(13)-108, C(9)-N(10)-H(14)-122, C(9)-N(10)-H(15)-118.
The atomic charges: C(1)-(0.511), C(2)-(-0.033), N(3)-(-0.394), C(4)-(0.03), C(5)-(0.418), N(6)-(-0.378), F(7)-(-0.283), O(8)-(-0.378), C(9)-(0.579), N(10)-(-0.527), O(11)-(-0.458), H(12)-(0.143), H(13)-(0.265), H(14)-(0.261), H(15)-(0.245) (see Fig. 1 ).
For model of favipiravir obtained by DFT method, the optimized bond lengths between carbon atoms in the ring are within the range of 1.39-1.51 Å, С-О-1.21-1.33 Å, С-Н-1.09 Å, C-N-1.30-1.35 Å, F-C-1.33 Å, H-N-1.00-1.01 Å, H-O-0.97 Å.
The following corresponding optimized bond angles (in degrees) were obtained: N(6)-C(2)-C(1)- 119,C(5)-N(6)-C(2)- 119, C(2)-C(1)-N(3)- 121, C(1)-N(3)-C(4)- 119, N(3)-C(4)-C(5)- 120, C(4)-C(5)-N(6)- 123, C(4)-C(5)-F(7)- 120, C(2)-C(1)-O(8)- 122, N(6)-C(2)-C(9)- 117, C(1)-C(2)-C(9)- 124, C(2)-C(9)-N(10)- 113, O(11)-C(9)-N(10)- 124, C(2)-C(9)-O(11)- 123, N(3)-C(4)-H(12)- 119, C(1)-O(8)-H(13)- 106, C(9)-N(10)-H(14)- 120, C(9)-N(10)-H(15)-118.
The atomic charges: C(1)-(0.416),C(2)-(-0.033), N(3)-(-0.347), C(4)-(-0.014),C(5)-(0.337), N(6)-(-0.357),F(7)-(-0.207), O(8)-(-0.294), C(9)-(0.439), N(10)-(-0.466), O(11)-(-0.361), H(12)-(0.148), H(13)-(0.249), H(14)-(0.25), H(15)-(0.24) (see Fig. 2 ).
Using formula pKa = 49.04 - 134.61 qmaxH+ [11], obtained by authors for AB INITIO/6-311G** (qmaxH+ = +0.265), we find the value pKa = 13.
For DFT-PBE0/6-311G** method the acidity index is calculated by formula pKa = 51.048-150.078qmaxH+ [7] (qmaxH+ = +0.25). Calculated value pKa = 14.
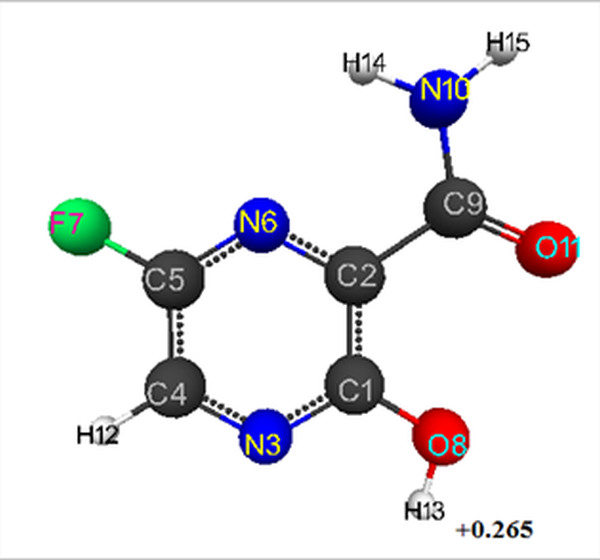
Figure 1. Geometric and electronic structure of favipiravir's molecule obtained by AB INITIO method.
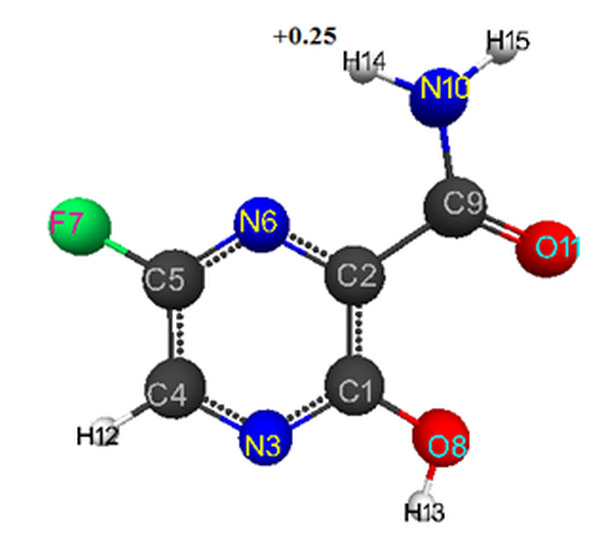
Figure 2. Geometric and electronic structure of favipiravir's molecule obtained by DFT method.
Table 1. Quantum-chemical parameters (E0, Eel, qmaxH+ and pKa) of favipiravir studied.
|
Preparation |
Method |
E0, kJ/mol |
Eel, kJ/mol |
qmaxH+ |
pKa |
|
Favipiravir |
AB INITIO |
-1586398 |
-3182282 |
+0.265 |
13 |
|
DFT |
-1593396 |
-3178870 |
+0.25 |
14 |
Thus, the optimized atomic-molecular structures of studied favipiravir were obtained by DFT and AB INITIO methods. At the same time, the quantum-chemical parameters (E0, Eel, qmaxH+), dipole moments, distribution of electron density on atoms were calculated, and also it was proved that studied favipiravir belongs to the class of weak acids (9 <pKa≤14). It is obvious that proposed hypothesis for dependence of favipiravir's efficiency on its acidic strength requires experimental verification.
References
- Fevipiravis. Dug Information Portal. U.S. National Library at Medicine.
- Avifavir. Directory of medicines. Medum.ru. 2016, 512 p. (in Russian).
- ru.wikipedia.org. Favipiravir [electronic resource] (in Russian).
- Quantum-chemical aspects of cationic polymerization of olefins, V.A. Babkin et al., 1996, Gilem Publishing House (Ufa), 188 pp. (in Russian).
- Granovsky, A. A., Firefly version 8, 2013. http://classic.chem.msu.su/gran/firefly/index.html
- MacMolPlt: A Graphical User Interface for GAMESS., B.M. Bode, M.S. Gordon, Journal of Molecular Graphics, 1998, 16, 133-138.
- General Atomic and Molecular Electronic Structure System, M.W. Schmidt et al., J.Comput.Chem. 1993, 14, 1347-1363.
- Quantum-Chemical Calculation of Some Molecules of Triftoromethylstyroles by the DFT Method. V.А. Babkin et al., Fluorine Notes, 2019, 123, 5-6
- Ermakov A.T. Quantum mechanics and quantum chemistry. Yurayt Publishing House, 2016, 555 p. (in Russian).
- Cirelson V.G., Quantum Chemistry. Molecules, molecular systems and solids, Moscow, Publishing House «Binom», 2010, 496 p. (in Russian).
- Connection of the universal acidity index of H-acid with the charge on Hydrogen atom (AB INITIO method). V. A. Babkin et al., Oxidation Communications, 2002, 25(1), 21-47.
ARTICLE INFO
Received 25 August 2020
Accepted 27 August 2020
Available online October 2020
Recommended for publication by Prof. S.M. Igumnov
Fluorine Notes, 2020, 132, 5-6
