Received: August 2020
DOI 10.17677/fn20714807.2020.04.04
Fluorine Notes, 2020, 131, 7-8
SYNTHESIS OF METHYL PERFLUOROALKYL- AND 1,1-DICHLOROPERFLUOROALKYL SULFIDES
A.A. Tyutyunovab, S. R. Sterlina, S. M. Igumnovab
a A.N.Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences,28 Vavilova St., 119991 Moscow, Russian Federation
bP&M-Invest Scientific Production Association,28 Vavilova St., 119991 Moscow, Russian Federation
e-mail: tuytuynov@rambler.ru
Abstract: The decarboxylation of alkali metals perfluorocarboxilates in the presence of methyl rhodanide in DMF leads to the formation of methyl perfluoroalkyl sulfides in 60-70% yields. The compounds obtained transform into methyl 1,1-dichloroperfluoroalkyl sulfides under the action of ClSO3H/CF3COOH in 60-65% yields.
Keywords: methyl perfluoroalkyl sulfides, chlorosulfonic acid, 1,1-dichloroperfluoroalkyl sulfides.
It is known that α,ω-bis(methylthio)perfluoroalkanes (C4-C10) are hydrolyzed with 98% H2SO4 at 150-180oC to give the corresponding perfluorinated carboxylic acids [1]. At the same time it is shown that hydrolysis of methyl-α-chloro-α-fluoroethyl sulfides is carried out under mild conditions at room temperature or heating for 10 min [2]. In turn methyl α-chloro- or α-bromoperfluoroalkyl sulfides in CH2Cl2 solution are either hydrolyzed under the action of more strong chlorosulfonic acid already at 0oC to give methyl thioesters or split with the formation of thioacyl halogenides [3]. It should be noted here that polyfluoroalkyl sulfides are split also under the action of Lewis acids with the formation of complicated unidentified mixture of products (AlCl3, SbF5) or thioacyl halogenides (TiF4, TiCl4) [3,4].
The difference in easiness of hydrolysis between lower and higher methyl perfluoroalkyl sulfides under the action of conc. H2SO4 is probably connected with their different solubility. Thus, methyl 4-H-octafluorobutyl sulfide hydrolyzes totally in the presence of conc. H2SO4 at 100oC/2 h. However methyl 8-H-hexadecafluorooctyl sulfide under the analogous conditions does not virtually react but when trifluoroacetic acid is added to the reaction mixture as a solvent hydrolysis is completed at 80oC within several hours.
Thereby alkyl perfluoroalkyl sulfides as well as their oxygen analogs can be used in practice for the synthesis of different perfluorinated carboxylic acids derivatives, and the choice in favour of ethers or thioethers depends on availability of the latter.
In order to synthesize methyl perfluorovinyloxy-acetate CF2=CFOCF2CO2CH3 - a demandable fluorocontaining monomer - we obtained a previously undescribed methyl perfluorovinyloxyethyl sulfide (2a) by modified method [5] (Scheme 1). In the course of investigation of sulfide 2a hydrolysis to perfluorovinyloxy-acetic acid it was found out that it is impossible to achieve selective hydrolysis of CF2S-moiety under the action of conc. H2SO4 or ClSO3H in hexane or methylene chloride medium as well as in presence of Lewis acids such as SbCl5, AlCl3, BBr3, BF3.EtO2.
In the course of our study we have found that the interaction of sulfide 2a with ClSO3H in trifluoroacetic acid leads to acid catalyzed nucleophylic substitution of fluorine atoms in α-position towards sulfur for chlorine with the formation of dichloride 3a and fluorosulfonic acid (Scheme 1). The carrying out of the given reaction with deficiency of ClSO3H (1.25 eq.) resulted in minimization of parallel reactions across CF2=CFO-group and allowed to isolate chloride 3a in ~40% yield (considering conversion 2a).
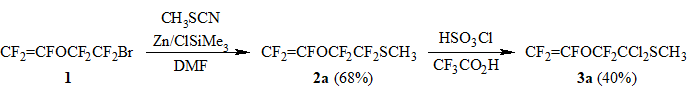
Scheme 1.
As such transformation of CF2S-moiety into CCl2S-group in perfluoroalkyl sulfides under the action of chlorosulfonic acid has not been described so far we studied the application of this reaction for the synthesis of different 1,1-dichloroperfluoroalkyl sulfides.
One of the most simple and available methods of perfluoroakyl sulfides synthesis is the approach developed
earlier that consists in decarboxylation of perfluorocarboxylic acids salts in the presence of disulfides
or rhodanides [6-9]. We investigated the possibility to apply methyl rhodanide [14] thereto because
its b.p. (130-131oC) allows to realize this reaction in an open system. It turned out
that decarboxylation of lower fluorocarboxylates CF3COONa or C2F5COONa
in the presence of CH3SCN resulted in the formation of sulfides 2b-c in ~10-20% yields. At that time methyl perfluoroalkyl sulfides were obtained in 60-70% yields by
the decarboxylation of higher perfluorocarboxylic acids salts in DMF (Scheme 2, Table 1).
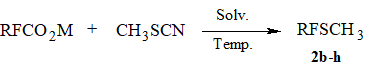
Scheme 2.
Table 1. Synthesis of methyl perfluoroalkyl sulfides 2b-h.
|
No |
RFCO2M |
Solv. |
Temp., oC |
Yield RFSCH3 (2b-h), % |
|
1 |
CF3CO2Na |
DMF |
140÷160 |
5 (2b) |
|
2 |
CF3CO2Na |
Sulfolane |
150÷180 |
10 (2b) |
|
3 |
CF3CO2Na |
DMF+FeCl2(30%) |
140÷160 |
6 (2b) |
|
4 |
C2F5CO2Na |
DMF |
140÷150 |
20 (2c) |
|
5 |
n-C3F7CO2Na |
Sulfolane |
150÷160 |
54 (2d) |
|
6 |
n-C3F7CO2K |
Sulfolane |
150÷160 |
55 (2d) |
|
7 |
n-C3F7CO2K |
Diglyme |
150÷160 |
50 (2d) |
|
8 |
n-C3F7CO2K |
Tetraglyme |
150÷160 |
56 (2d) |
|
9 |
n-C3F7CO2K |
DMF |
140÷150 |
69 (2d) |
|
10 |
n-C3F7CO2Na |
DMF |
140÷150 |
70 (2d) |
|
11 |
n-C3F7CO2K |
PhCN |
160÷170 |
47 (2d) |
|
12 |
n-C3F7CO2Na |
NMP |
140÷150 |
58 (2d) |
|
13 |
n-C3F7CO2Na |
DMF+CuCl(5%) |
140÷150 |
46 (2d) |
|
14 |
n-C3F7CO2Na |
DMSO |
140÷150 |
62 (2d) |
|
15 |
n-C3F7CO2K |
DMA |
140÷150 |
65 (2d) |
|
16 |
n-C5F11CO2Na |
DMF |
140÷150 |
70 (2e) |
|
17 |
H(CF2)4CO2Na |
DMF |
140÷150 |
63 (2f) |
|
18 |
H(CF2)8CO2Na |
DMF |
140÷150 |
60 (2g) |
|
19 |
n-C6F13CO2Na |
DMF |
140÷150 |
65 (2h) |
The reaction of sulfides 2b-g odtained by the given method with chlorosulfonic acid
in CF3COOH medium is realized similarly to that described above with the formation of
methyl 1,1-dichloroperfluoroalkyl sulfides 3b-f (Scheme 3).
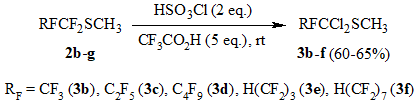
Scheme 3.
Traditional treatment of halogen exchange in α-halogensulfides under the action of halogen hydrides assumes the intermediate formation of sulfonium halides as a result of sulfur atom protonation [10-11].
Polyhalogenated dimethyl sulfides are also capable to enter the halogen exchange reaction under the action of halogen hydrides, but in these cases it is more probable that protonation is directed at halogen atoms instead of sulfur atom which electron donating properties are descended under the influence of electron acceptor substituents that allows to consider such exchange as nucleophylic substitution with electrophilic assistance [12-13].
Everything mentioned above refers equally to the interaction of methyl perfluoroalkyl sulfides with HSO3Cl/CF3COOH. The fluorine atoms in α-position towards sulfur atom possess definite anionoid mobility and at the first stage of the reaction are protonated with the formation of HF and the product of fluorine substitution for O-anion (trifluoroacetate or chlorosulfonyloxy-group). Hydrogen fluoride reacts readily with chlorosulfonic acid to give fluorosulfonic acid and HCl – the source of chloride-anion, that successfully competes with other anions in acid catalyzed substitution of fluorine atoms.
The anionoid mobility of fluorine is evidently a key factor in the exchange reactions of α-fluorine atoms in fluoroaliphatic sulfides. This property manifests especially brightly in reactivity of diperfluoroalkyl disulfides in which CF2-groups adjacent to disulfide moiety hydrolyze totally in aq. THF or dioxane with intermediate formation of the corresponding diacyl disulfides (NMR 19F) (we failed to isolate analytical samples as these compounds hydrolyze readily to give the carboxylic acids). The further workup of the reaction products with H2SO4 led to the formation of the corresponding carboxylic acids.
Thus in the course of our study a simple procedure of higher methyl perfluoroalkyl sulfides preparation has been developed, it was shown that these compounds react with ClSO3H in CF3COOH medium to give methyl-1,1-dichloroperfluoroalkyl sulfides and demonstrated the easiness of diperfluoroalkyl disulfides hydrolysis with the formation of the corresponding carboxylic acids.
Experimental
1H, 19F, and 31P NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300, 282, and 121 MHz, accordingly; the external standard was CDCl3. Chemical shifts for 1H spectra are presented vs. the residual signal of the solvent (δ 7.26) and are given in ppm vs. tetramethylsilane. Chemical shifts in 19F spectra are given in ppm vs. CFCl3. Downfield shifts are positive. Chemical shifts in 31P spectra are given vs. 85% H3PO4. Mass spectra are recorded using a Finnigan Polaris Q mass spectrometer (Trace GC ultra). Elemental analysis was carried out in Laboratory of Microanalysis of A.N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences.
Methyl perfluoro-2-vinyloxyethyl sulfide (2a).
Trimethylsilyl chloride (9 g) is added to a suspension of zinc powder (112 g, 1.7 g-at) in 600 ml of DMF, the mixture is stirred for 10-15 min, then under vigorous stirring a mixture of 2-bromoperfluoroethyl vinyl ether (338 g, 1.22 mol), methyl rhodanide (107 g, 1.46 mol), Me3SiCl (6 g) and DMF (100 ml) is added with the rate that sustains the reaction temperature at 25-30oC (the temperature is controlled by the rate of addition and external cooling), after the addition is over the mixture is stirred for 3h at 25-30oC, the volatile products are distilled off at reduced pressure (10 Torr) into cooled receiver (-40÷-30oC) collecting the fraction up to 40÷45oC. The distillate obtained is washed with equal volume of 5% aq. HCl, distilled over equal volume of conc. H2SO4 and purified by rectification to give 210 g (68%) of 2a, purity 99%, b.p. 101oC.
Found, %: C 24.44; H 1.49; F 54.10; S 13.10. C5H3F7OS.
Calc., %: C 24.60; H 1.24; F 54.48; S 13.13.
NMR 1H, δ: 2.6 (s, CH3);
NMR 19F, δ: -137,6 (dd, 1F, 3JFF-trans = 112 Hz, 3JFF-cis = 68 Hz, OCF), -125,6 (dd, 1F, 2JFF = 90 Hz, =CF-trans), -118,1 (dd, 1F, 2JFF = 90 Hz, =CF-cis), -97,2 (с, 2F, SCF2), -89,5 (c, 2F, OCF2).
Methyl 1,1-dichloroperfluoro-2-vinyloxyethyl sulfide (3a).
Chlorosulfonic acid (35 g, 0.3 mol) is added dropwise under stirring to a cooled (10oC) mixture of sulfide 2a (60 g, 0.24 mol) and 140 ml of CF3COOH. The reaction mixture is stirred for several hours and stayed for 24 h at 15-20oC. Then the reaction mixture is poured on crashed ice, the organic layer is separated, washed with water, mixed with equal volume of 25% aq. NH3, stirred for 10-15 min, separated and distilled at 10 Torr over P2O5. The distillate obtained is rectified to isolate starting compound 2a and target compound 3a. There is obtained 21 g of dichlorosulfide 3a (yield 40% considering conversion 2a), b.p. 161.5oC, that contains 7% of byproduct CF3CFHOCF2CCl2SCH3.
NMR 1H, δ: 2.4 (s, CH3);
NMR 19F, δ: -136,9 (dd, 1F, 3JFF-trans = 112 Hz, 3JFF-cis = 68 Hz, OCF), -124,8 (dd, 1F, 2JFF = 90 Hz, =CF-trans), -117,2 (dd, 1F, 2JFF = 90 Hz, =CF-cis), -81,7 (c, 2F, OCF2).
Decarboxylation of CF3CO2Na in the presence of CH3SCN in DMF.
The mixture of sodium trifluoroacetate (51.85 g, 0.38 mol), methyl rhodanide (30.1 g, 0.41 mol) and 140 ml of DMF is heated under stirring for 2 h, increasing gradually the temperature of the reaction mixture from 140oC to 170oC with simultaneous stripping of reaction products collecting the fraction with b.p. up to ~80oC. There is obtained 13.75 g of a mixture that contains mainly CF3SCH3 (δ: -45) и CF3CO2Me (δ: -76) in molar ratio 1:3 (according to NMR 1H, NMR 19F and CMS-data). The yield of methyl trifluoromethyl sulfide does not exceed 10%.
The decarboxylation of C3F7CO2Me in the presence of methyl rhodanide in different solvents (Table 1).
The solution of potassium or sodium perfluorobutyrate (0.08 mol) and 6.4 g of methyl rhodanide in 50 ml of solvent is heated on oil bath with simultaneous stripping of the reaction products into receiver (-78oC) for 2 h. The distillate obtained is analyzes by NMR 1H, NMR 19F and GLC.
General procedure of methyl perfluoroalkyl sulfides 2c-h synthesis.
The mixture of sodium perfluorocarboxylate (0.33 mol), 30 g (0.41 mol) of methyl rhodanide and 200 ml of DMF is heated under stirring on oil bath for 2 h (until gas evolution ceases) with simultaneous stripping of the reaction products into receiver (-78oC) supplied with reflux condenser (-78oC) connected with Tishchenko flask with conc. H2SO4. In the case of high boiling sulfides the target product is distilled off together with DMF under reduced pressure (10 Torr). The distillate obtained is washed with diluted hydrochloric acid and distilled over 50 vol.% of conc. H2SO4 collecting the product in receiver (-78oC). The product is purified by rectification. Yield of 2c ~20%, yields of 2d-h 60-65%.
Methyl perfluoroethyl sulfide (2c): b.p. 37-38oС (lit. 36.5оС [2]).
NMR 1H, δ: 2.6 (s, CH3);
NMR 19F, δ: -97.8 (s, 2F, SCF2), -85,9 (s, 3F, CF3).
Methyl perfluoropropyl sulfide (2d): the purity of sulfide 2d after rectification is 95% (the impurity substance is methyl perfluoro-i-propyl sulfide), b.p. 64-65оС (lit. 61оС [15]).
NMR 1H, δ: 2.6 (s, CH3);
NMR19F, δ: -126.5 (s, 2F, CF2), -93.8 (m, 2F, SCF2), -82,8 (s, 3F, CF3).
Methyl perfluoroamyl sulfide (2e): b.p. 111÷111.5оС.
NMR 1H, δ: 2,4 (с, CH3);
NMR 19F, δ: -128.4+-124.3+-122.2 [s, 2F+2F+2F, (CF2)3], -93,1 (m, 2F, SCF2), -83.6 (m, 3F, CF3).
Methyl 4-H-perfluorobutyl sulfide (2f): b.p. 116-117оС.
NMR 1H, δ: 2.5 (s, 3H, CH3), 6,1 (tt, 1H, 2JHF = 51Hz, 3JHF = 4,8 Hz, HCF2);
NMR 19F, δ: -139.9 (d, 2F, 2JHF = 51Hz, HCF2), -131.8+-123.9 [s, 2F+2F, (CF2)2], -92.8 (s, 2F, SCF2).
Methyl 8-H-perfluorooctyl sulfide (2g): b.p. 74-74.5оС/10 Торр.
Found, %: C 24.12; H 1.13; F 67.73; S, 7.00. C9H4F16S.
Calc, %: C 24.12; H 0.90; F 67.83; S, 7.15.
NMR 1H, δ: 2.3 (s, 3H, CH3), 5.9 (tt, 1H, 2JHF = 51 Hz, 3JHF = 4.8 Hz, HCF2);
NMR 19F δ: -139.9 (d, 2F, 2JHF = 51Hz, HCF2), -132.1+-125.5+-123.9+-123.3+-122.0 [s, 2F+2F+4F+2F+2F, (CF2)6], -93,1 (m, 2F, SCF2).
Methyl perfluorohexyl sulfide (2h): b.p. 133-134оС (lit. 131÷132оС [16]).
NMR 1H, δ: 2.4 (s, CH3);
NMR 19F, δ: -128.4+-124.8+123.5+-122.1 [s, 2F+2F+2F+2F, (CF2)4], -93.1 (m, 2F, SCF2), -83,7 (m, 3F, CF3).
General procedure of methyl 1,1-dichloroperfluoroalkyl sulfides 3b-f synthesis.
Chlorosulfonic acid (114 g, 0.4 mol) is added to a solution of sulfide 2b-g (0.2 mol) in CF3COOH (114 g, 1 mol) at 10oC under stirring. The reaction mixture is stayed overnight, poured on crashed ice, lower layer is separated, washed with water and distilled over P2O5. The target product is purified by rectification.
Methyl 1,1-dichloro-2,2,2-trifluoroethyl sulfide (3b): b.p. 113.5-114оС (lit. 57оС/100 Торр) [2]). NMR 1H, δ: 2.4 (s, CH3);
NMR 19F, δ: -78.5 (s, CF3).
Methyl 1,1-dichloroperfluoropropyl sulfide (3c): b.p. 132.5оС.
NMR 1H, δ: 2.4 (s, CH3);
NMR 19F, δ: -112.2 (s, 2F, CF2CCl2), -77.6 (m, 3F, CF3).
Methyl 1,1-dichloroperfluoroamyl sulfide (3d). b.p. 54.5-55оС/10 Торр.
NMR 1H, δ: 2.4 (s, CH3);
NMR 19F, δ: -128 (м, 2F, CF2), -118.5 (s, 2F, CF2), -107.8 (s, 2F, CF2CCl2), -83 (m, 3F, CF3).
Methyl 1,1-dichloro-4-H-perfluorobutyl sulfide (3e). b.p. 55.5оС/10 Торр.
NMR 1H, δ: 2.35 (s, 3H, CH3), 5.9 (tt, 1H, 2JHF = 52 Hz, 3JHF = 4.8 Hz, HCF2);
NMR 19F, δ: -138.6 (d, 2F, 2JFF = 52Hz, HCF2), -125.9 (c, 2F, CF2), -108.8 (s, 2F, CF2CCl2).
Mass-spectrum (M/Z, reference): 281[M+H]+, 261[M-F]+, 245[M-Cl]+, 233[M-SCH3]+, 94[ClCSCH3]+, 79[ClCS]+(100%), 67[SCl]+, 63[CF2CH]+, 59[C2Cl]+, 51[CF2H]+, 47[SCH3]+, 45[CHS]+.
Methyl 1,1-dichloro-8-H-perfluorooctyl sulfide (3f). b.p. 70оС/0,5 Торр.
Found, %: C 22.39; H 0.99; Cl 14.60; F 55.36; S 6.74. C9H4Cl2F14S.
Calc., %: C 22.47; H 0.84; Cl 14.74; F 55.29; S 6.67.
NMR 1H, δ: 2,3 (s, 3H, CH3), 5.75 (tt, 1H, 2JHF = 52 Hz, 3JHF = 4,8 Hz, HCF2);
NMR 19F, δ : -139.5 (d, 2F, 2JFF = 52Hz, HCF2), -131.8+-125.3+-123.5+-117.5 [s, 2F+2F+4F+2F, (CF2)5], -107,8 (c, 2F, CF2CCl2).
Acknowledgments
This work was performed with the financial support from Ministry of Science and higher Education of the Russian Federation using the equipment of Center for molecular composition studies of INEOS RAS.
References
- R.B.Ward, J.Org.Chem., 1965, 30, 3009-3011.
- R.C.Terrell, T.Ucciardi, J.F.Vitcha, J.Org.Chem., 1965, 30, 4011-4013.
- T.Nguyen, C.Wakselman, J.Fluor.Chem., 1987, 35, 523-530.
- K.E.Rapp, J.T.Barr, R.L.Pruett, C.T.Bahner, J.D.Gibson, R.H.Lafferty Jr., J.Am.Chem.Soc., 1952, 74, 749-753.
- M.Tordeux, C.Francese, C.Wakselman, J.Fluor.Chem., 1989, 43, 27-34.
- B.Quiclet-Sire, R.N.Saicic, S.Z.Zard, Tetrahed.Lett., 1996, 37, 9057-9058.
- N.Roques J.Fluor.Chem., 2001, 107, 311-314.
- B.Exner, B.Bayarmagnai, F.Jia, L.J.Goossen, Eur.J.Org.Chem., 2015, 21, 17220-17223.
- B.Exner, B.Bayarmagnai, C.Matheis, L.J.Goossen, J.Fluor.Chem., 2017, 198, 89-93.
- F.Boberg, G.Winter, G.R.Schultze, Chem.Ber., 1956, 89, 1160-1169.
- Yu.V.Pokonova. Haloid sulfides (Methods of preparation, properties, application of haloidthioethers). Ed. of Leningrad Univercity, Leningrad, 1977. (In Russian)
- F.Boberg, G.Winter, G.R.Schultze, Liebigs Ann.Chem., 1959, 621, 8-19.
- L.Saint-Jalmes, J.Fluor.Chem., 2006, 127, 85-90.
- W.V. Rochat, G.L. Gard J.Org.Chem., 1969, 34, 4173-4176.
- R.N.Haszeldine, B.Higginbottom, R.B.Rigby, A.E.Tipping, J.Chem.Soc.,Perkin Trans. 1, 1972, 155-159.
- R.N.Haszeldine, A.E.Tipping, Patent DE 2238458 (1973).
ARTICLE INFO
Received 06 August 2020
Accepted 17 August 2020
Available online August 2020
Recommended for publication by PhD M. Manaenkova
Fluorine Notes, 2020, 131, 7-8
