Received: February 2020
DOI 10.17677/fn20714807.2020.01.03
Fluorine Notes, 2020, 128, 5-6
SYNTHESIS AND REACTIONS OF PERFLUOROALKANE-α-HYDRO- AND -α,α-DIHYDROSULFONYL BROMIDES WITH ALKENES
A.A. Tyutyunovab, L.F. Ibragimovaa
a A.N.Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences,28 Vavilova St., 119991 Moscow, Russian Federation
bPiM-Invest Scientific Production Association,28 Vavilova St., 119991 Moscow, Russian Federation
e-mail: tuytuynov@rambler.ru
Abstract: Synthetic methods to obtain perfluoroalkane-α-hydro- and -α,α-dihydrosulfonyl bromides have been elaborated. Reactivity of these compounds towards allyl acetate and other aliphatic alkenes was studied. It was found that perfluoroalkane-α-hydrosulfonyl bromides add to alkenes predominantly with the elimination of SO2, while the corresponding α,α-dihydrosulfonyl bromides afford sulfones in the same reaction.
Keywords: difluoromethanesulfonyl bromide, perfluoroalkane-α-hydrosulfonyl bromides, perfluoroalkane-α,α-dihydrosulfonyl bromides, 2,2,2-trifluoroethanesulfonyl bromide, fluoroalkylation.
Successful application of trifluoromethanesulfonyl bromide as a trifluoromethylating agent in the production of high-demand organofluorine compounds such as 5,5,5-trifluoropentan-1-ol, 4,4,4-trifluorobut-2-en-1-ol, 4,4,4-trifluorobutylamine, 2-(2,2,2-trifluoroethyl)oxirane, etc. [1] as well as a series of fluorinated carboranes [2] where using of other fluoroalkylating agents is less effective, arouses interest in further investigations in the chemistry of fluoroalkanesulfonyl bromides. The aim of the present study was to synthesize hitherto unknown α-hydro- and α,α-dihydrofluoroalkanesulfonyl bromides and to study their properties.
Unlike aliphatic sulfonyl bromides whose addition to unsaturated compounds generates the corresponding sulfones [3-10], their perfluorinated analogues generally react with alkenes and acetylenes with the elimination of SO2 [11-20], with a few exceptions [1]. Therefore, it was of special interest for us to elucidate the influence of the hydrogen atom in the α-position to SO2Br-group on the reactivity of fluorinated sulfonyl bromides towards alkenes.
Perfluoroalkane-α-hydrosulfonyl bromides 1a-e were obtained by a standard procedure via the ring-opening reaction of available perfluoroalkane-β-sultones 2a-e [21-22] with water or HCl [23-25] yielding the corresponding sulfonyl fluorides 3a-e or chlorides 4c-e with their subsequent transformation into sulfinates and bromination according to the study [9].
Scheme 1
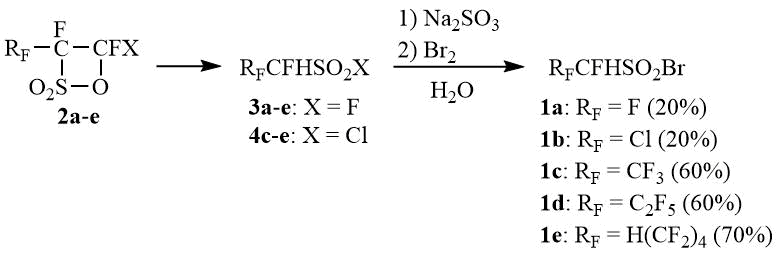
This procedure can be easily scaled up and enables the synthesis of perfluoroalkane-α-hydrosulfonyl bromides in favourable yields except for difluoromethanesulfonyl bromide (1a) and its chlorine analogue 1b, for which the ring-opening of sultones 2a-b proceeds to give sulfonyl fluorides 3a-b with the yields of as low as ~20% [23, 26].
All synthesized α-hydrosulfonyl bromides 1a-e are stable compounds, slowly releasing SO2 when exposed to sunlight to afford the corresponding bromides (the conversion of compounds 1a-e in sunlight within 2 days is 18 (within 7.5 h), 2, 0, 11, and 100%, respectively). The rate of desulfonylation rises significantly with the elongation of the perfluoroalkyl radical or upon addition of acetonitrile (e.g., compounds 1b-c quantitatively transform into bromides in sunlight upon addition of MeCN within 2-3 h). Thermal decomposition of 1a-e is observed at temperatures >130°C. Thus, α-hydrosulfonyl bromides 1a-e are thermally and photochemically more stable than their perfluorinated analogues.
Reactivity of α-hydrosulfonyl bromides 1a-e towards alkenes was studied using allyl acetate as a model substrate (5), which shows medium reactivity in such reactions.
It was found that difluoromethanesulfonyl bromide (1a) and chlorofluoromethanesulfonyl bromide (1b) convert to bromodifluoromethane (6a) and bromochlorofluoromethane (6b), respectively, in the presence of a three-fold molar excess of freshly distilled allyl acetate at room temperature within several days in the absence of direct sunlight. A polymer product is concurrently formed, representing presumably polysulfone 7.
Scheme 2
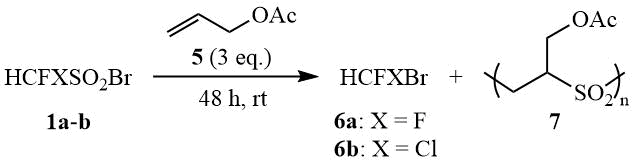
Carrying out the above reaction in sunlight or in acetonitrile solution in the presence of metals (Cu, Fe, Mn, Co) also results in the formation of bromides 6a-b.
It is worth noting that trifluoromethanesulfonyl bromide is almost unreactive under the same conditions, however being photo-, thermally or chemically activated, CF3SO2Br adds to allyl acetate with the elimination of SO2 [1, 15].
Cyclohexene and 1-decene are more reactive towards sulfonyl bromides and react with compounds 1a-b similar to allyl acetate yielding bromides 6a-b along with minor amounts of unidentified adducts. Taking into account the amphiphilic character of HCF2 radical, we examined reaction of substrates 1a-b with certain alkenes bearing electron-withdrawing substituents (ethylacrylate, acrylonitrile), however in this case compounds 1a-b are also transformed into bromides 6a-b and give substantially no products of the addition across the C=C bond.
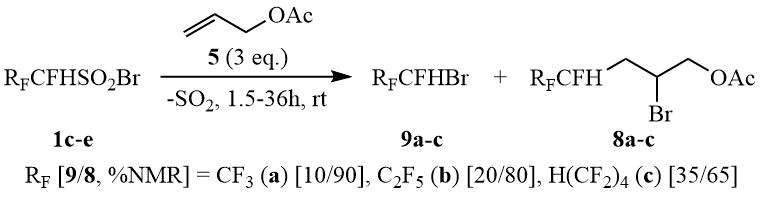
At the same time in the reaction of higher α-hydrosulfonyl bromides 1c-e with allyl acetate without direct sunlight at 20°C the release of SO2 is observed and adducts 8a-c are formed in good yields. The addition rate increases noticeably along with elongation of the perfluoroalkyl radical, however the same tendency is observed for the yields of side products i.e. bromides 9a-c. When carrying out this reaction in sunlight, along with the products 8a-c and 9a-c, a considerable amount of a polymer, presumably polysulfone 7, is formed.
Scheme 3
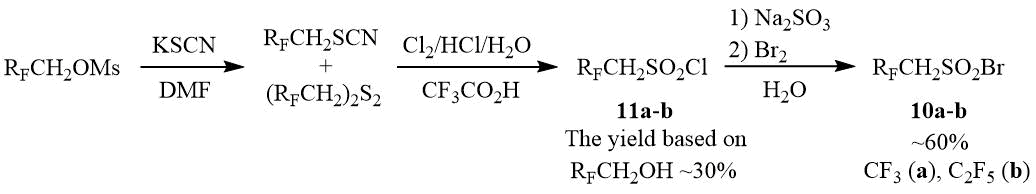
Perfluoroalkane-α,α-dihydrosulfonyl bromides 10a-b were synthesized according to the same approach including the transformation of relatively available sulfonyl chlorides 11a-b [27-28] to sulfinates followed by bromination.
Scheme 4
Alternatively, α,α-dihydrosulfonyl bromides can be obtained from the corresponding sulfonyl fluorides derived from β-sultones of 2-H-perfluoroalkenes [29].
Scheme 5
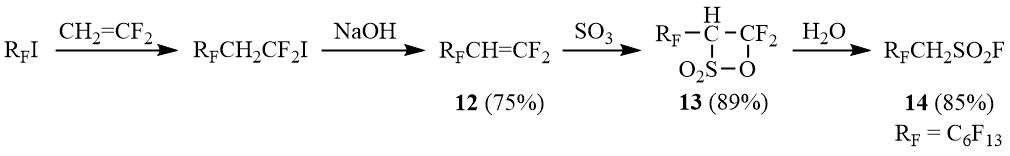
α,α-Dihydrosulfonyl bromides 10a-b are stable compounds. Being exposed to sunlight, they undergo desulfonylation slower than α-hydrosulfonyl bromides even in the presence of acetonitrile (compound 10a transforms into the corresponding bromide under sunlight in the presence of MeCN within 5 h in 8% yield).
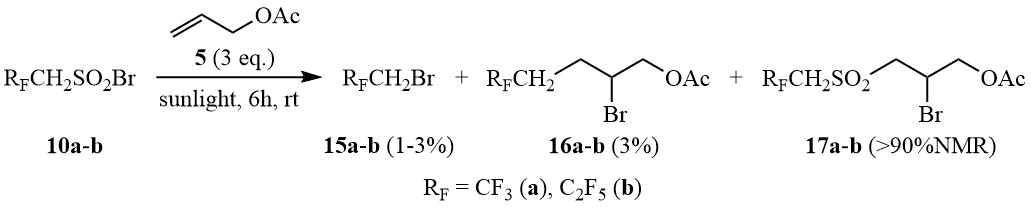
Studying the reaction between hydrosulfonyl bromides 10a-b and allyl acetate demonstrated that in the presence of sunlight the addition furnishes the corresponding sulfones 17a-b.
Scheme 6
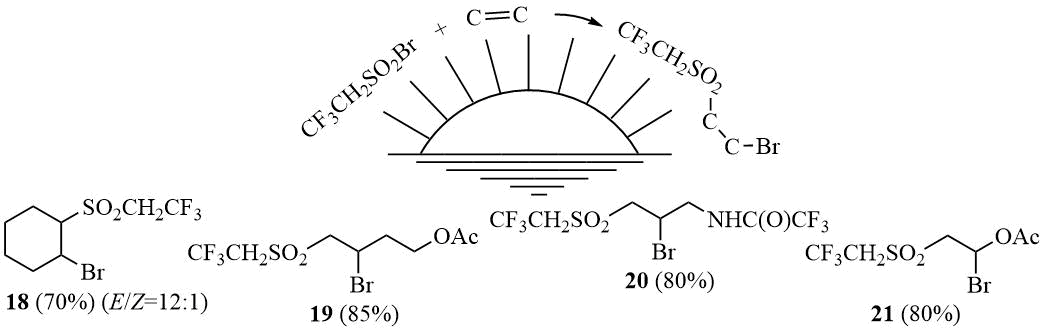
The same reaction behaviour towards compound 10a under sunlight is also observed for more reactive alkenes such as cyclohexene and homoallyl acetate, their reactions being completed in a few hours, as well as less active N-allyltrifluoroacetamide, which adds to this hydrosulfonyl bromide within 3-5 days depending on the sunlight intensity. The addition products formed with the elimination of SO2 as well as bromide 15a are generated in minor amounts in these reactions.
Scheme 7
In conclusion, we have shown that perfluoroalkane-α-hydro- and -α,α-dihydrosulfonyl bromides can be used as perfluoroalkylating agents. α-Hydrosulfonyl bromides were found to add to alkenes predominantly with the elimination of SO2, while α,α-dihydrosulfonyl bromides behave in this reaction similar to non-fluorinated sulfonyl bromides affording sulfones. In particular, it was shown that CF3CH2SO2Br adds smoothly to donor alkenes in the presence of sunlight and is a convenient reagent for the synthesis of a variety of compounds bearing 2,2,2-trifluoroethanesulfonyl group.
Experimental
1H, 19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300 and 282 MHz, accordingly; the external standard was CDCl3. Chemical shifts for 1H spectra are presented vs. the residual signal of the solvent ( δ 7.26) and are given in ppm vs. tetramethylsilane. Chemical shifts in 19F spectra are given in ppm vs. CFCl3. Downfield shifts are positive. Mass spectra are recorded using a Finnigan Polaris Q mass spectrometer (Trace GC ultra). Elemental analysis was carried out in Laboratory of Microanalysis of A.N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences.
The compounds used in this work are commercially available or obtained according to the methods described in literature: difluoromethanesulfonyl fluoride (3a) [23], chlorofluoromethanesulfonyl fluoride (3b) [30] and 2,2,2-trifluoroethanesulfonyl chloride (11a) [28, 30].
Synthesis of sulfonyl fluorides 3c-e (general procedure) [25].
A mixture of sultone 2c-e (0.2 mol) and 150 ml of water was stirred at 20÷30°C for several hours until gas evolution ceased; the lower layer was separated and distilled over P2O5.
Synthesis of sulfonyl chlorides 4c-e (general procedure) [24].
To ethereal solution of dry HCl [36.5 g (1 mol) in 250 ml of dry ether] under stirring at -20÷-10°C (0.2 mol) of sultone 2c-e is added; the resulting mixture was stirred at 20÷25°C for 1 hour and left overnight. Then, the solvent was distilled off via Vigreux column, the residue was washed with cold water until decarboxylation ceased, separated and distilled over P2O5. The obtained product was additionally purified by rectification.
1H-Perfluoropropanesulfonyl chloride (4d). b.p. 10°C/10 torr. 1H NMR δ: 6.1 (dd, 1H, 2JHF = 44 Hz, 3JHF = 18 Hz, CF2CFH); 19F NMR δ: -181 (m, 1F, CFH), -123, -130.1 (ABq, 2F, 2JFF = 288 Hz, CF2CFH), -84 (d, 3F, 3JFF = 11 Hz, CF3).
1H-Perfluoropentanesulfonyl chloride (4e). b.p. 105°C/10 torr. 1H NMR δ: 5.95 (dd, 1H, 2JHF = 42 Hz, 3JHF = 18 Hz, CF2CFH), 6.05 (t, 1H, 2JHF = 48 Hz, HCF2); 19F NMR δ: -180 (m, 1F, CFH), -139.5 (m, 2F, HCF2), -131 (m, 2F, HCF2CF2), -125.4 (m, 2F, HCF2CF2CF2), -120.7, -125.7 (ABq, 2F, 2JFF = 296 Hz, CF2CFH).
Synthesis of sulfonyl bromides 1a-e (general procedure).
To a solution of 41.6 g (0.33 mol) of Na2SO3 in 200 ml of water the sulfonyl chloride 4c-e (0.15 mol) was added dropwise under stirring at 5÷10°C or sulfonyl fluoride 3a-e at 20°C. Then the resulting mixture was stirred at 25÷30°C until total dissolution of sulfonyl chloride 4c-e (1-2 hours) or at 35÷55°C in the case of reactions with sulfonyl fluorides 3a-e (about 10% acetonitrile was added to the reaction mixture in order to accelerate the reaction with higher sulfonyl fluorides). Then, bromine (48 g, 0.3 mol) was added dropwise under stirring at -5÷0°C until a stable bromine color of the reaction mixture appeared. The lower layer was separated and distilled over P2O5 at 10-0.5 torr. The product obtained was purified by rectification.
Difluoromethanesulfonyl bromide (1a). b.p. 31÷32°C/16 torr. 1H NMR δ: 6.3 (t, 2JHF = 53 Hz, HCF2); 19F NMR δ: -113.7 (d, HCF2).
Chlorofluoromethanesulfonyl bromide (1b). b.p. 63.5÷64.5°C/19 torr. Found (%): C, 5.57; H, 0.41; Br, 38.18; Cl, 16.64; F, 8.99; S, 15.03. CHBrClFO2S. Calculated (%): C, 5.68; H, 0.48; Br, 37.79; Cl, 16.77; F, 8.99; S, 15.16. 1H NMR δ: 6.6 (d, 2JHF = 48 Hz, HCFCl); 19F NMR δ: -126.5 (d, HCFCl).
1H-Perfluoroethanesulfonyl bromide (1c). b.p. 30÷31оС/10 torr. Found (%): C, 9.87; H, 0.47; Br, 33.16; F, 30.98. C2HBrF4O2S. Calculated (%): C, 9.81; H, 0.41; Br, 32.62; F, 31.02. 1H NMR δ: 5.8 (dq, 1H, 2JHF = 44 Hz, CFH); 19F NMR δ: -179 (m, 1F, CFH), -73 (m, 3F, CF3).
1H-Perfluoropropanesulfonyl bromide (1d). b.p. 45°C/10 torr. Found (%): C, 12.30; H, 0.36; Br, 28.21; F, 38.21. C3HBrF6O2S. Calculated (%): C, 12.21; H, 0.34; Br, 27.09; F, 38.64. 1H NMR δ: 5.9 (dd, 1H, 2JHF = 42 Hz, 3JHF = 18 Hz, CF2CFH); 19F NMR δ: -178 (m, 1F, CFH), -123, -129.5 (ABq, 2F, 2JFF = 288 Hz, CF2CFH), -84 (d, 3F, 3JFF = 11 Hz, CF3).
1H,5H-Perfluoropentanesulfonyl bromide (1e). b.p. 53°C/0.5 torr. Found (%): C, 14.80; H, 0.45; Br, 22.56; F, 45.13. C5H2BrF9O2S. Calculated (%): C, 15.93; H, 0.53; Br, 21.19; F, 45.35. 1H NMR δ: 5.8 (dd, 1H, 2JHF = 45 Hz, 3JHF = 15 Hz, CF2CFH), 6.0 (t, 1H, 2JHF = 45 Hz, HCF2); 19F NMR δ: -177 (m, 1F, CFH), -139 (m, 2F, HCF2), -131 (m, 2F, HCF2CF2), -125 (m, 2F, HCF2CF2CF2), -120.8, -124.6 (ABq, 2F, 2JFF = 293 Hz, CF2CFH).
Reactions of difluoro- and chlorofluoromethanesulfonyl bromides (1a-b) with allyl acetate (5).
A mixture of difluoro- or chlorofluoromethanesulfonyl bromide (1a-b) (7.7 mmol) and 2.3 g (23.1 mmol) of allyl acetate (5) was kept at 20÷25°C in 30 ml tube for 2 days in the absence of direct sunlight. In the course of reaction the formation of white dense polymer precipitate was observed. According to 19F NMR data, the solution contains bromodifluoromethane (6a) (δ: -69.5, d, 2JHF = 62 Hz) or bromochlorofluoromethane (6b) (δ: -82, d, 2JHF = 52 Hz).
Bromodifluoromethane (6a). 1H NMR δ: 7.6 (t, 2JHF = 60 Hz, HCF2); 19F NMR δ: -68.7 (d, HCF2).
Bromochlorofluoromethane (6b). 1H NMR δ: 8.2 (d, 2JHF = 52 Hz, HCF); 19F NMR δ: -82 (d, HCFClBr).
The polymer precipitate was separated, washed with a mixture of acetone and ethyl acetate (1:1) and dried in a vacuum of 0.1 torr. After grinding, a white powder of polysulfone 7 was obtained. Found (%): C, 38.77; H, 5.33; S, 17.32. C5H8O4S. Calculated (%): C, 36.58; H, 4.91; S, 19.53. IR (KBr, cm-1) max: 600, 641, 734, 848, 1049, 1130, 1228, 1324, 1371, 1750, 2947, 3001.
Reactions of sulfonyl bromides 1c-e with allyl acetate (5).
A mixture of sulfonyl bromide 1c-e (0.01 mol) and 3 g (0.03 mol) of allyl acetate (5) was kept at 20÷25°C in 30 ml tube for 1.5 days in the absence of direct sunlight.
The reaction time depends on the amount of peroxides present in allyl acetate that are formed during its storage [31]. If the concentration of peroxides is high a vigorous exothermic reaction accompanied with gas evolution is observed in a minute after mixing of reagents, the temperature of the mixture rises to 70÷80°C and the formation of side products is registered. The rectified (immediately before the reaction) allyl acetate reacts with 1c-d for 1-1.5 days and with 1e for 1.5 hour under heating. It can be assumed that reaction is initiated by peroxides formed due to the presence of oxygen. When the reaction is carried out with freshly rectified 5 the formation of side polymer impurities is practically not observed. According to 19F NMR, the mixture contains 10-35% bromide 9a-c and 90-65% of adduct 8a-c. The volatile components of this mixture were distilled off under reduced pressure at 25÷50°C/15-0.5 torr into trap (-78°C), to give adducts 8a-c as distillation residue, which were isolated by distillation in a vacuum.
2-Bromo-4,5,5,5-tetrafluoropentyl acetate (8a). Mixture of stereoisomers, b.p. 90÷91°C/10 torr. 1H NMR δ: 2.0 (s, 3H, CH3), 2.3÷2.5 (m, 1H, CHBr), 4.3 (m, 4H, CH2CHBrCH2), 5.1 (dq, 1H, 2JHF = 48 Hz, CFH); 19F NMR δ: -205 + -200 (m, 1F, CFH), -81 (m, 3F, CF3). The mass spectrum (M/Z, reference): 281[M+H]+, 220[C5H5BrF4]+, 200[C5H4BrF3]+, 181[C5H4BrF2]+, 141[C5H5F4]+, 121[C5H4F3]+, 101[CF3CFH]+, 95[C3H2F3]+, 77[C3H3F2]+, 69[CF3]+, 51[CHF2]+, 43[CH3CO]+(100%).
2-Bromo-4,5,5,6,6,6-hexafluorohexyl acetate (8b). Mixture of stereoisomers, b.p. 90÷91°C/10 torr. 1H NMR δ: 2 (s, 3H, CH3), 2.4÷2.6 (m, 1H, CHBr), 4.3 (m, 4H, CH2CHBrCH2), 5.2 (dq, 1H, 2JHF = 48 Hz, CFH); 19F NMR δ: -206 + -201 (m, 1F, CFH), -134.3, -125.9 + -133.5, -126.6 (ABq, 2F, 2JFF = 285 Hz, CF2), - 84 (m, 3F, CF3). The mass spectrum (M/Z, reference): 331[M]+, 272[C6H6BrF6]+, 270[C6H4BrF6]+, 250[C6H3BrF5]+, 191[C6H5F6]+, 171[C6H4F5]+, 151[C3HF6]+, 145[C4H2F5]+, 127[C4H3F4]+, 113[C3HF4]+, 101[C2HF4]+, 77[C3H3F2]+, 69[CF3]+, 51[CHF2]+, 43[CH3CO]+(100%).
2-Bromo-4,5,5,6,6,7,7,8,8-nonafluorooctyl acetate (8c). Mixture of stereoisomers, b.p. 90÷92°C/0.5 torr. 1H NMR δ: 1.9 (s, 3H, CH3), 2.3÷2.5 (m, 1H, CHBr), 4.3 (m, 4H, CH2CHBrCH2), 5.2 (dq, 1H , 2JHF = 48 Hz, CFH), 6.1 (t, 1H, 2JHF = 47 Hz, HCF2); 19F NMR δ: -206 + -201 (m, 1F, CFH), -139 (m, 2F, HCF2), -131 (m, 2F, HCF2CF2), -125 (m, 2F, HCF2CF2CF2), -129.6, -123.4 + -128.7, -123.8 (ABq, 2F, 2JFF = 291 Hz, CF2CFH). The mass spectrum (M/Z, reference): 413[M]+, 354[C8H7BrF9]+, 352[C8H5BrF9]+(100%), 332[C8H4BrF8]+, 273[C8H6F9]+, 253[C8H5F8]+, 227[C6H3F8]+, 209[C6H4F7]+, 183[C4H2F7]+, 163[C4HF6]+, 151[C4H5BrF]+, 145[C4H2F5]+, 113[C3HF4]+, 95[C3H2F3]+, 69[CF3]+, 51[CHF2]+, 43[CH3CO]+.
2,2,3,3,3-Pentafluoropropanesulfonyl chloride (11b). Obtained analogously to 11a, b.p. 51÷52°C/10 torr. Found (%): C, 15.57; H, 0.82; Cl, 15.25; F, 40.85; S, 13.76. C3H2ClF5O2S. Calculated (%): C, 15.49; H, 0.87; Cl, 15.24; F, 40.85; S, 13.79. 1H NMR δ: 4.6 (t, 2H, 3JHF = 15 Hz, CH2); 19F NMR δ: -118 (t, 2F, 3JHF = 15 Hz, CF2), -87 (s, 3F, CF3).
2,2,2-Trifluoroethanesulfonyl bromide (10a). To a solution of 214 g (1.7 mol) of Na2SO3 in 800 ml of water, 155 g (0.85 mol) of 2,2,2-trifluoroethanesulfonyl chloride (11a) were added under stirring at 10°C. The mixture was stirred at 15°C for 1.5 hours, cooled to 5°C, and 272 g (1.7 mol) of bromine were added dropwise until a bromine color appeared. The lower layer was separated, distilled over P2O5 in a vacuum and further purified by rectification. Yield 60%, b.p. 51.5°C/10 torr. Found (%): C, 10.51; H, 0.99; Br, 35.23; F, 25.17. C2H2BrF3O2S. Calculated (%): C, 10.58; H, 0.89; Br, 35.20; F, 25.11. 1H NMR δ: 4.7 (q, 2H, 3JHF = 7 Hz, CH2); 19F NMR δ: -62 (t, 3F, CF3).
2,2,3,3,3-Pentafluoropropanesulfonyl bromide (10b). Obtained analogously to 10a, b.p. 60°C/10 torr. 1H NMR δ: 4.7 (t, 2H, 3JHF = 14 Hz, CH2); 19F NMR δ: -117 (t, 2F, 3JHF = 14 Hz, CF2), -87 (s, 3F, CF3).
1,1,3,3,4,4,5,5,6,6,7,7,8,8,8-Pentadecafluorooct-1-ene (12). A mixture of 150 g (0.294 mol) of C6F13CH2CF2I and 17.6 g (0.44 mol) of NaOH in 150 ml of sulfolane was heated under vigorous stirring until a vigorous exothermic reaction began. The mixture was kept under reflux for 10-15 minutes and the reaction products were distilled off. Further distillation over P2O5 afforded 84 g (75% yield) of 12 containing 10% C5F11CF=CHCF3. The product 12 obtained was used without further purification. b.p. 105÷110°C. 1H NMR δ: 4.2 (td, 2H, 3JHF = 14 Hz, CH2); 19F NMR δ: -127+-125+-124+-123 (s, 2F+2F+2F+2F, CF2CF2CF2CF2), -110 (s, 2F, CF2CH=CF2), -83 (t, 3F, 3JFF = 7.5 Hz, CF3), -74.5 (d, 1F, 3JFF = 11 Hz, CH=CFcisFtrans), -74.5 (d, 1F, 4JFF = 11 Hz, CH=CFcisFtrans).
2-Hydroperfluorooctan-β-sultone (13). A mixture of 136 g (0.356 mol) of olefin 12 and 31.2 g (0.39 mol) freshly distilled sulfuric anhydride was heated in a steel swinging autoclave at 70÷75°C for 5 hours. The autoclave was opened, the contents were heated to 50÷60°C, put off into a distillation flask and distilled at 15 torr to give 146 g of 13 (yield 89%), colorless crystals, m.p. 56÷57°C, b.p. 106÷108оС/15 torr. 1H NMR δ: 4.2 (td, 2H, 3JHF = 14 Hz, CH2); 19F NMR δ: -127+-123+-122 (s, 2F+4F+2F, CF2CF2CF2CF2), -113 (d, 2F, 4JFF = 11 Hz, CF2CH2SO2F), -82 (d, 3F, 3JFF = 8.5 Hz, CF3), 65 (t, 1F, 4JFF = 11 Hz, SO2F).
2H,2H-Perfluoroheptanesulfonyl fluoride (14). The reaction of 13 with warm water (similarly to 3c-e [29]) gave sulfonyl fluoride 14, m.p. 56÷57°C, b.p. 86°C/10 torr. Found (%): C, 20.34; H, 0.49; F, 63.83; S, 7.78. C7H2F14O2S. Calculated (%): C, 20.20; H, 0.48; F, 63.92; S, 7.70. 1H NMR (Freon 113) δ: 4.2 (td, 2H, 3JHF = 14 Hz, CH2); 19F NMR (Freon 113) δ: -127+-123+-122 (s, 2F+4F+2F, CF2CF2CF2CF2), -113 (d, 2F, 4JFF = 11 Hz, CF2CH2SO2F), -82 (d, 3F, 3JFF = 8.5 Hz, CF3), 65 (t, 1F, 4JFF = 11 Hz, SO2F).
Reactions of sulfonyl bromides 10a-b with allyl acetate (5).
A mixture (0.01 mol) of sulfonyl bromide (10a or 10b) and 3 g (0.03 mol) of allyl acetate (5) was kept at 20÷25°C in a 30 ml tube in the sunlight on a windowsill for 6 hours. According to 19F NMR data the mixture contained ~1-3% bromides 15a-b (15a δ: -70, t, 3JHF = 8.5 Hz, CF3), ~3% of adducts 16a-b (16a δ: -66.9, t, 3JHF = 11 Hz, CF3), and >90% of adducts 17a or 17b correspondingly. The volatile components of the reaction mixture were distilled off in a vacuum at 25÷50°C/15-0.5 torr into a trap (-78°C), to give adducts 17a-b as residue.
2-Bromo-3-[(2,2,2-trifluoroethyl)sulfonyl]propyl acetate (17a). b.p. 130÷135°C/0.5 torr. 1H NMR δ: 2.1 (s, 3H, CH3), 3.8 (br.d, 2H, CH2OAc), 3.9÷4.2 (m, 2H, CF3CH2), 4.37, 4.46 (dABq, 2H, 2JHH = 12 Hz, 3JHH = 4 Hz, SO2CH2CHBr), 4.6 (quint, 1H, CHBr); 19F NMR δ: -61.6 (t, 3F, 3JHF = 8.5 Hz, CF3). The mass spectrum (M/Z, reference): 327[M+H]+, 266[M-CH3CO2H]+, 247[M-Br]+, 203[M-HBr-CH3CO]+, 187[M-HBr-CH3CO2]+, 147[CF3CH2SO2]+, 119[CH2CBrCH2]+(100%), 99[C4H3SO]+, 83[CF3CH2]+, 67[C4H3O]+, 43[CH3CO]+, 39[C3H3]+.
2-Bromo-3-[(2,2,3,3,3-pentafluoropropyl)sulfonyl]propyl acetate (17b). b.p. 130÷133°C/0.5 torr. 1H NMR δ: 2.1 (s, 3H, CH3), 3.8 (br.d, 2H, CH2OAc), 3.8÷4.2 (m, 2H, CF3CH2), 4.37, 4.45 (dABq, 2H, 2JHH = 12 Hz, 3JHH = 10 Hz, SO2CH2CHBr), 4.6 (quint, 1H, CHBr); 19F NMR δ: -117 (t, 2F, 3JHF = 17 Hz, CF2), -86 (t, 3F, 3JFF = 11 Hz, CF3). The mass spectrum (M/Z, reference): 377[M+H]+, 357[M-F]+, 319[C6H7O2SBrF5]+, 297[M-Br]+, 277[C8H9O4SF4]+, 237[C6H6O2SF5]+, 217[C6H4O2SF4]+, 179[C5H8O2Br]+, 119[C3H4Br]+, 99[C4H7O2]+, 69[CF3]+, 43[CH3CO]+(100%).
(E) 1-Bromo-2-[(2,2,2-trifluoroethyl)sulfonyl]cyclohexane (18). A mixture of 10.2 g (0.045 mol) of sulfonyl bromide 10a and 11 g (0.13 mol) of cyclohexene was loaded in a 30 ml tube and placed on a windowsill. As a result of solar illumination the exothermic reaction began with evolution of small amounts of SO2. The mixture was kept on a windowsill during the day. The volatile components of this mixture were distilled off in a vacuum at 0.5 torr into a trap affording adduct 18 as distillation residue. Further distillation gave 9.7 g (70%) of adduct 18, the E:Z-isomers ratio = 12:1, b.p. 125÷130°C/0.5 torr. 1H NMR δ: 1.4+1.75+1.95+2.45 (m, 2H+2H+2H+2H, Cy), 3.45, 3.95 (m, 1H+1H, CF3CH2), 4.4 (m, 2H, CHCySO2+CHCyBr); 19F NMR δ: -61.1 (t, 3F, 3JHF = 8.5 Hz, CF3). The mass spectrum (M/Z, reference): 309[M+H]+, 289[M-F]+, 229[M-Br]+, 225[M-CF3CH2]+, 209[M-CF3CH2O]+, 161[M-CF3CH2SO2]+, 149[C5H10Br]+, 119[C3H4Br]+, 94[C7H10]+, 81[C6H9]+(100%), 79[Br]+, 68[C5H8]+, 53[C4H5]+, 39[C3H3]+.
2-Bromo-4-[(2,2,2-trifluoroethyl)sulfonyl]butyl acetate (19). The adduct 19 was obtained similarly to 17a, b.p. 130÷135°C/0.5 torr. Found (%): C, 27.91; H, 3.52; F, 16.57; S, 9.42. C8H12BrF3O4S. Calculated (%): C, 28.17; H, 3.55; F, 16.71; S, 9.40. 1H NMR δ: 2.1 (s, 3H, CH3), 2.15+2.4 (m, 1H+1H, CHBrCH2), 3.7+3.9+4.2+4.3 (m, 1H+2H+2H+1H, CF3CH2SO2CH2CHBrCH2CH2OAc), 4.5 (m, 1H, CHBr); 19F NMR δ: -61.6 (t, 3F, 3JHF = 8.5 Hz, CF3). The mass spectrum (M/Z, reference): 340[M]+, 281[M-CH3CO2]+, 261[M-Br]+, 241[M-Br-HF]+, 217[M-HBr-CH3CO]+, 201[M-HBr-CH3CO2]+, 149[C6H7F2S]+, 133[C4H6Br]+, 83[CF3CH2]+, 69[CF3]+, 53[C4H5]+(100%), 43[CH3CO]+, 39[C3H3]+.
N-(2-Bromo-4-[(2,2,2-trifluoroethyl)sulfonyl]butyl)-2,2,2-trifluoroacetamide (20). A mixture of 7 g (0.03 mol) of sulfonyl bromide 10a and 9.4 g (0.06 mol) of N-allyltrifluoroacetamide is maintained at 15÷20°C in a 30 ml tube in sunlight on a windowsill for 3-5 days (depending on the intensity of sunlight), during which a crystalline product was formed. The reaction mixture was diluted with equal volume of CH2Cl2, the precipitate was filtered off, washed with CH2Cl2 and dried to give 4.6 g (80% taking into account ~50% conversion) of adduct 20, m.p. 116÷117°C. Found (%): C, 22.25; H, 2.22; F, 29.99; N, 3.69. C7H8BrF6NO3S. Calculated (%): C, 22.12; H, 2.12; F, 29.99; N, 3.69; S, 8.43. 1H NMR (DMSOd6) δ: 3.7 (m, 2H, CH2NH), 3.9÷4.1 (m, 2H, CF3CH2), 4.6 (m, 1H, CHBr), 4.7 (m, 2H, SO2CH2CHBr), 9.7 (br.s, 1H, NH); 19F NMR (DMSOd6) δ: -76.4 (s, 3F, C(O)CF3), -61.3 (t, 3F, 3JHF = 8.5 Hz, CF3CH2).
1-Bromo-2-[(2,2,2-trifluoroethyl)sulfonyl]ethyl acetate (21). The adduct 21 was obtained by addition of 2,2,2-trifluoroethanesulfonyl bromide (10a) to vinyl acetate under conditions similarly to the preparation of 17a. 1H NMR (CDCl3) δ: 2.1 (s, 3H, CH3), 3.9+4.1 (m, 3H+1H, CF3CH2SO2CH2), 6.9 (m, 1H, CHBr); 19F NMR (CDCl3) δ: -61.5 (t, 3F, 3JHF = 8.5 Hz, CF3). The mass spectrum (M/Z, reference): 313[M+H]+, 293[M-F]+, 273[M-F-HF]+, 255[M-CH3CO-CH2]+, 233[M-Br]+, 43[CH3CO]+(100%).
Acknowledgments
This study was financially supported by the Ministry of Science and Higher Education of the Russian Federation with the use of scientific equipment of the Centre of Molecular Structure Research of the INEOS RAS.
References
- A.A. Tyutyunov, L.F. Ibragimova, N.D. Kagramanov, S.R. Sterlin, S.M. Igumnov, Fluorine Notes, 2016, 109, 1-2.
- V.A. Ol’shevskaya, A.A. Tyutyunov, I.F. Ibragimova, E.G. Kononova, E.G. Rys, Polyhedron, 2019, 171, 508-514.
- S.J. Cristol, D.I. Davies, J.Org.Chem., 1964, 29, 1282-1284.
- L.I. Zakharkin, G.G. Zhigareva, Zh.Org.Khim., 1973, 9, 891-895.
- W. Böll, US 4022804 (1977).
- W. Böll, Lieb.Ann.Chem., 1979, 1665-1674.
- E. Block, M. Aslam, J.Am.Chem.Soc., 1983, 105, 6164-6165.
- E. Block, M. Aslam, V. Eswarakrishnan, A. Wall J.Am.Chem.Soc., 1983, 105, 6165-6167.
- E. Block, M. Aslam, V. Eswarakrishnan, K. Gebreyes, J. Hutchinson, R. Iyer, J.-A. Laffitte, A. Wall, J.Am.Chem.Soc., 1986, 108, 4568-4580.
- A.S. Gozdz, P. Maslak, J.Org.Chem., 1991, 56, 2179-2189.
- W.-Y. Huang, J.-L. Chen, L.-Q. Hu, Bull.Soc.Chim.Fr., 1986, 6, 881-884.
- W.-Y. Huang, J.-L. Chen, Acta Chim.Sinica, Eng.Ed., 1988, 150-154.
- Y.-F. Zhang, L. Lu, W.-Y. Huang, ActaChim.Sinica, Eng.Ed., 1989, 376-384.
- W.-Y. Huang, H.-Z. Zhang, Chin.J.Chem., 1991, 9, 76-83.
- W.-Y. Huang, L. Lu, Chin.J.Chem., 1992, 10, 268-273.
- W.-Y. Huang, H.-Z. Zhang, Chin.J.Chem., 1992, 10, 274-277.
- W.-Y. Huang, H.-Z. Zhang, Chin.J.Chem., 1992, 10, 544-548.
- X.-K. Jiang, G.-Z. Ji, J.R.-Y. Xie, J.Fluor.Chem., 1996, 79, 133-138.
- Patent CN 1730127 (2010).
- A.A. Tyutyunov, L.F. Ibragimova, N.D. Kagramanov, N.I. Delyagina, V.F. Cherstkov, S.R. Sterlin, S.M. Igumnov, Fluorine Notes, 2015, 102, 1-2.
- I.L. Knunyants, G.A. Sokolski, Angew.Chem.Int.Ed., 1972, 11, 583-595.
- J. Mohtasham, G.L. Gard, Coord.Chem.Rev., 1992, 112, 47-79.
- G.A. Sokol’skii, I.L. Knunyants, Bull.Acad.Sci.USSR, Div.chem.sci., 1961, 10, 1499-1501.
- G.A. Sokol’skii, M.A. Belaventsev, I.L. Knunyants, Bull.Acad.Sci.USSR, Div.chem.sci., 1967, 16, 1471-1474.
- L.N. Ragulin, P.P. Ropalo, G.A. Sokol’skii, I.L. Knunyants, Bull.Acad.Sci.USSR, Div.chem.sci., 1967, 16, 1685-1688.
- Q.-Y. Chen, S.-W. Wu, J.Fluor.Chem., 1990, 47, 509-514.
- R.E. Oesterling, US 3006964 (1961).
- R.K. Crossland, W.E. Wells, V.J. Shiner Jr., J.Am.Chem.Soc., 1971, 93, 4217-4219.
- N.P. Aktaev, G.A. Sokol’skii, I.L. Knunyants, Bull.Acad.Sci.USSR, Div.chem.sci., 1975, 24, 2416-2421.
- Sintezy Ftororganicheskikh Soedinenii. (Syntheses of Organofluorine Compounds). Pt. 3-4. Eds S.M. Igoumnov, E.V. Igoumnova. Moscow: NPO “PiM-Invest”, 2015, 2019. (in Russian)
- M.S. Kharasch, F.R. Mayo, J.Am.Chem.Soc., 1933, 55, 2468-2496.
ARTICLE INFO
Received 07 February 2019
Accepted 18 February 2020
Available online February 2020
Recommended for publication by Prof. S. Sterlin
Fluorine Notes, 2020, 128, 5-6
