Received: December 2019
DOI 10.17677/fn20714807.2019.06.04
Fluorine Notes, 2019, 127, 7-8
CHLOROMETHYLATION OF POLYFLUOROAROMATIC COMPOUNDS
M.B. Saporovskaya1, V.E. Boiko1,2, V.L.Don1,2, S.M. Igumnov1,2
1A.N.Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilova st.,28, Moscow, 119991 Russia
2 "P&M-Invest" Scientific Industrial Association, Ltd., Vavilova st., 28, Moscow, 119991 Russia
e-mail: veronika.don@gmail.com
Abstract: Chloromethylation of a number of polyfluoroaromatic compounds is described, including 2,3,5,6-tetrafluorobenzene to produce both mono-and bis-chloromethylation products depending on the reaction conditions, pentafluorobenzene, 2,4,6-trifluoro (trifluoromethyl) benzene, and others, as well as chloromethylation of difluorobenzene to produce mono- and bis-chloromethyldifluorobenzene. Further transformations of the resulting chloromethyl derivatives to produce fluoroaromatic compounds of various classes are described.
Кeywords:.Chloromethylation, bis(chloromethyl) ether, polyfluoroaromatic compound, chloromethyl polyfluoroaromatics, pentafluorobenzyl chloride, tetrafluorobenzyl chloride, 3,5-Bis(chloromethyl)-2,4,6-trifluorobenzotrifluoride
Chloromethylation of aromatic compounds is a synthetically important reaction, that opens the way
for the synthesis of a number of aromatic derivatives - methyl, hydroxymethyl, formyl and others.
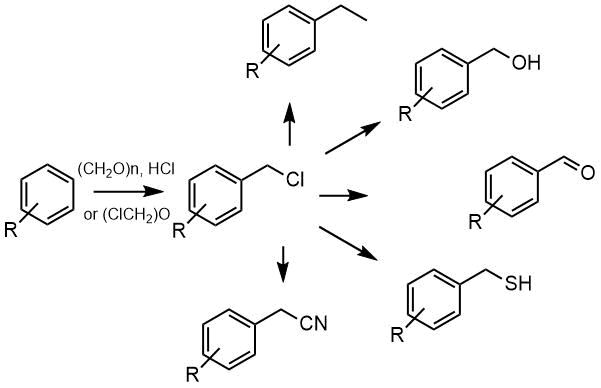
Since then in 1898 Grasse and Mosely succeeded in replacing a hydrogen atom in benzene with a chloromethyl group by the reaction of benzene with paraform in hydrogen chloride in the presence of zinc chloride, this reaction has been actively studied; a number of reviews have been devoted to aromatic chloromethylation [1,2].
Since chloromethylation is electrophilic reaction, electron-rich aromatics react under milder conditions, usually in an aqueous medium by treatment with formaldehyde in any form, HCl, and aluminum or zinc chloride [1]. For chlormethylation of deactivated aromatic compounds, pre-prepared chloromethyl or bis (chloromethyl) ethers are mainly used [3], or a chloromethylating mixture is prepared from paraform, chlorosulfonic and concentrated sulfuric acids [4].
The presence of halogens in the aromatic ring makes it difficult to introduce a chloromethyl group; polyhalogenated aromatic compounds are easier to involve in this reaction if they contain electron-donating alkyl or hydroxy groups along with halogens, [5,6].
Chloromethylation of polymers, in particular polystyrenes and aromatic polyesters, has been widely developed; the products obtained are used in particular for the preparation of membranes for fuel cells [7-9].
As for fluorinated aromatic compounds, in addition to chloromethylation of monofluorobenzene, chloromethylation of difluorobenzene with chloromethyl ether in a mixture of acetic anhydride with sulfuric acid with a yield of 42-45% is described [10]. Chloromethylation of 1,3,5-trifluorobenzene with paraform in a mixture of acetic and sulfuric acids with zinc chloride and gaseous HCl [11], as well as chloromethylation of 2,3,5,6-tetrafluorotoluene with a yield of 85% are described [3]. In the Chinese patent [4] chloromethylation of 1,2,5,6-tetrafluorobenzene using a mixture of sulfuric and chlorosulfonic acids, zinc chloride and paraform is disclosed, but the yields of chloromethylation products are not given. Patent [13] describes the chloromethylation of 3,4,5-trifluorobromobenzene to give a monomethylated product in 80% yield.
We carried out chloromethylation of some fluorinated benzenes.
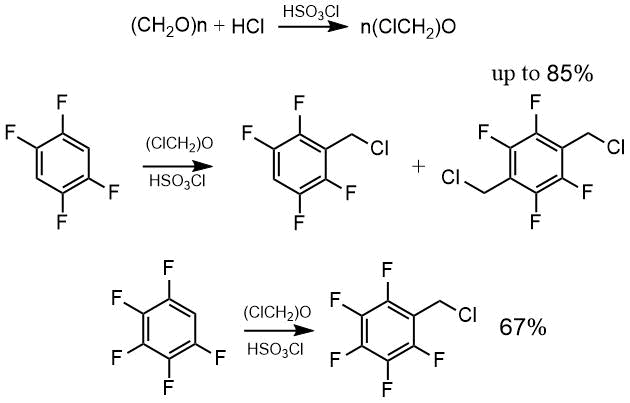
Our attempt to chloromethylate 2,3,5,6-tetrafluorobenzene by passing hydrogen chloride while heating it with paraform in the presence of zinc chloride did not lead to the formation of a product.
Using crude bis (chloromethyl) ether in chlorosulfonic acid, we obtained both mono- and bis-chloromethylation products of 2,3,5,6-tetrafluorobenzene. To obtain the product of mono-chloromethylation of tetrafluorobenzene, the reaction was carried out at the lower temperature (40°C), however, chloromethyltetrafluorobenzene was obtained in 27% yield, or 46% at a conversion of about 60%, while at the higher temperatures (70-100°C) a product of bis-chloromethylation was isolated in the yield up to 85%.
Under the same conditions (70-100°C), chloromethylpentafluorobenzene was prepared from pentafluorobenzene in 67% yield.
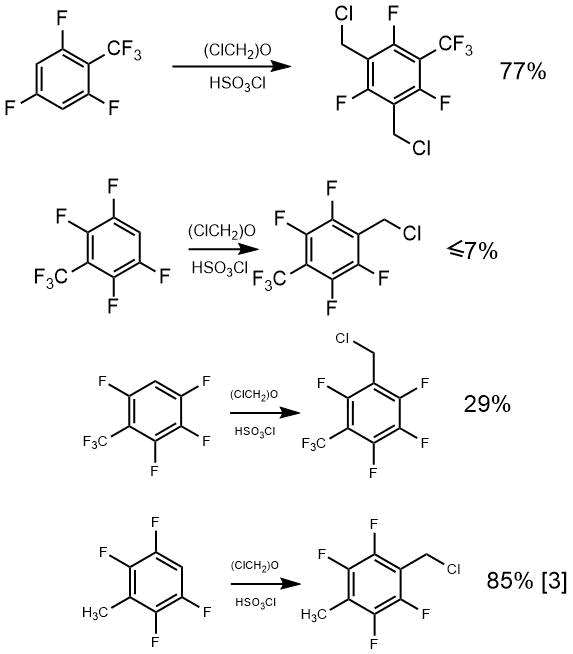
3,5-Bis(chloromethyl)-2,4,6-trifluorobenzotrifluoride was obtained from 2,4,6-trifluoro(trifluoromethyl) benzene in 77% yield. Chloromethylation of 2,3,5,6-tetrafluorobenzotrifluoride under similar conditions only led to traces of the desired product (about 7%), hydrogen in metha- position to trifluoromethyl groop of 2,4,5,6-tetrafluorobenzotrifluoride was replaced with a chloromethyl group in 29% yield, while 2,3,5,6-tetrafluorotoluene gave the product of chloromethylation in 85% yield, as described previously by Shteingarts [3].
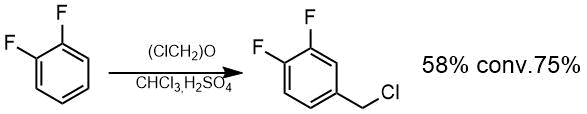
An attempt to chloromethylate difluorobenzene under the same severe conditions did not allow us to isolate the chloromethyl derivative because polymer products were formed mainly. Bis-chloromethylated product was obtained predominantly when difluorobenzene reacts with bis(chloromethyl) ether in sulfuric acid under cooling. Mono-chloromethylation product –chloromethyl-3,4-difluorobenzene was obtained in chloroform at a temperature not higher than room temperature in 58% yield, taking into account 75 % conversion.
The resulting chloromethyl derivatives were used further at synthesis, thus hydrolysis of chloromethyl groop with potassium carbonate in water or acetolysis followed by hydrolysis gave corresponding alcohols, chlorination of chloromethyl group gave benzalchlorides and trichloromethylbenzenes, and further corresponding aldehydes and acids, benzyl bromides were obtained by the reaction with an aqueous solution of sodium bromide in the presence of triethyl(benzyl)ammonium chloride (TEBAC) [14a-c].
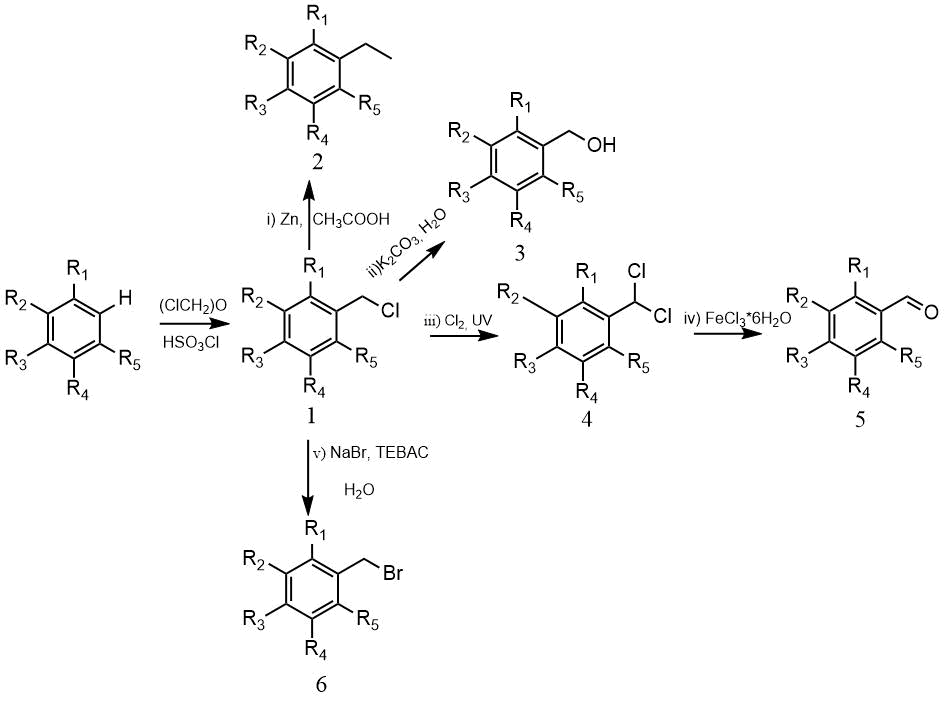
(1-6) (a) 1=R2=R3=R4=R5=F, (b) R1=R2= R4=R5=F, R3=H, (c) R1=R2= R4=R5=F, R3=CH3,
(d) R1=R2= R4=R5=F, R3=CF3, (e) R1=R3= R4= R5=F, R2=CF3,
i). Zn, CH3COOH. 2b - yield 75%, B.p. 125-126°С, 2c–yield 88%, 2d– yield 80%.
By the reduction of 1,4-bis(chloromethyl)tetrafluorobenzene 1,4-bis(methyl)tetrafluorobenzene (m.p. 34-35°С, B.p. 145-147°С) was isolated.
ii). K2CO3, H2O 3c–yield 77%, M.p. 60-62°С, 3d –yield 78%, b.p.168-169°С, m.p.45-48°С.
iii). Cl2, UV, 4a - 57% with a conversion of 66%, b.p. 78°С /30 mm Hg.
iv). FeCl3*6H2O, 5a– yield 96% , b.p. 68-70°С/15 mm Hg.
v). NaBr, TEBAC, H2O, 6a–yield 68%, (or 83% with a conversion of 76%), b.p. 174-175°С.
Experimental
1H, 19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300 and 282 MHz, accordingly; the external standard was CDCl3. Chemical shifts for 1H spectra are recorded relative to the residual signal of the solvent (CHCl3 δ: 7.26) and are provided in ppm relative to TMS. Chemical shifts in 19F spectra are given in ppm relative to CFCl3. Downfield shifts are positive.
Bis(chloromethyl) ether [15]
Conc. hydrochloric acid (217 g , 184 ml) and paraform (268 g) were placed into a 2 l four-necked flask, immersed in an ice bath and provided with a mechanical stirrer, thermometer, dropping funnel and reflux condenser connected at the outlet to Tishchenko’s flask with conc. H2SO4. The contents of the flask were cooled in an ice water bath and chlorosulfonic acid (496 ml) was added dropwise while stirring, keeping the temperature below 10°C. Then the reaction mixture was allowed to warm to room temperature, stirred for 4 hours and left overnight. The upper layer, 460 g of bis(chloromethyl) ether, was separated and used in the next step without further purification.
2,3,4,5,6-Pentafluorobenzyl chloride (1a)
To pentafluorobenzene (200 g, 1,19 mol) and bis(chloromethyl) ether (76 g, 0,66 mol) while stirring and cooling to a temperature of 0-2°C, chlorosulfonic acid (117 g, 1,00 mol) was added dropwise at such a rate that the temperature does not rise above 5°C, after which the mixture was allowed to warm to room temperature over an hour, then gradually heated for two hours to 100°C and stirred at this temperature for about 1.5 hours until the evolution of HCl ended. The reaction mixture was cooled, carefully poured onto crushed ice. The organic layer was separated, washed with water until neutral, dried with MgS04 and distilled under reduced pressure to yield 172 g of 2,3,4,5,6-pentafluorobenzyl chloride 99% purity. B.p. 87°С/60 mm Hg. Yield 67%.
19F NMR (δ, ppm): -146,1 (m, 2F), -156,8 (m, 1F), -165,3 (m, 2F).
1H NMR (, ppm): 4,4 (s, 2H).
2,3,5,6-Tetrafluorobenzyl chloride (1b)
To 1,2,4,5-tetrafluorobenzene (567 g, 3,78 mol) and bis(chloromethyl) ether (491 g, 4,27 mol) while stirring and cooling to a temperature of 0–5°C, chlorosulfonic acid (432 ml, 3,7 mol) was added dropwise at such a rate that the temperature does not rise above 5°C, and HCl evolution does not be too vigorous, then the mixture was gradually brought to room temperature over an hour, stirred at this temperature for an additional hour, then slowly heated to 40°C, stirred at this temperature for 3 hours and allowed to stay overnight.* The reaction mixture was cooled, carefully poured onto crushed ice with methylene chloride (1,5 l). The organic layer was separated, washed with water until neutral, dried with MgS04. The solvent was distilled off, then a fraction, boiling in a temperature range 90-100°С, containing mainly the initial product, was distilled off. The residue was distilled under reduced pressure, collecting a fraction, boiling 56-65°С/10 mm Hg. As a result of a second distillation 2,3,5,6-tetrafluorobenzyl chloride (200 g) of 97% purity with b.p. 60°С/10 mm Hg. was obtained (27% yield or 46% taking into account 60% conversion).
19F NMR (, ppm) :-140,4 (m, 2F), -145,4 (m, 2F).
1H NMR (, ppm): 4,3 (s, 2H), 6,7 (m, 1H).
*At higher temperatures the yield of 1,4-bis(chlorometyl)tetrafluorobenzene instead of 2,3,5,6-tetrafluorobenzyl chloride increases.
1,4-Bis(chloromethyl) tetrafluorobenzene (1c)
According to the procedure similar to that described for 2,3,4,5,6-pentafluorobenzyl chloride (1a) from 1,2,4,5-tetrafluorobenzene (249 g), of bis(chloromethyl) ether (380 g) and chlorosulfonic acid (250 ml) was obtained 1,4-bis(chloromethyl)tetrafluorobenzene (1c) (252 g, 98+ purity). B.p. 105-110°С /15 mm Hg , m.p. 77-78С. Yield 67%.*
19F NMR (, ppm):-140,2 (m, 4F).
1H NMR (, ppm): 4,3 (s, 2H).
*When the light fraction of the product, which is a mixture of chloromethyl-tetrafluorobenzene with the initial tetrafluorobenzene was re-chloromethylated, the yield can be increased to 84%.
3,5-Bis(chloromethyl)-2,4,6-trifluorobenzotrifluoride (1g)
According to the procedure similar to that described for 2,3,4,5,6-pentafluorobenzyl chloride (1a), from 2,4,6-trifluorobenzotrifluoride (22 g, 0,11 mol), bis(chloromethyl) ether (30 g) and chlorosulfonic acid (18 ml) 3,5-bis(chloromethyl)-2,4,6-trifluorobenzotrifluoride (24 g, purity 98% ) was obtained in 77% yield. B.p. 130°С /45 mm Hg.
19F NMR (, ppm):-58,5 (m, 3F), -110,5 (m, 1F), -112,6 (m, 2F).
1H NMR (, ppm): 4,3 (s, 2H).
2,4,5,6-Tetrafluoro-3-(trifluoromethyl)benzyl chloride (1f)
According to the procedure similar to that described for 2,3,4,5,6-pentafluorobenzyl chloride (1a), from 2,4,5,6-tetrafluorobenzotrifluoride (280 г, 1,28 mol), bis(chloromethyl) ether (225 g, 1,96 mol) and chlorosulfonic acid (395 g, 3,39 mol) 2,4,5,6-tetrafluoro-3-(trifluoromethyl)-benzyl chloride (1f) (100 g, 0,375 mol) was obtained in 29% yield, b.p. 95°С /10 mm Hg,
19F NMR (, ppm): -59,2 (m, 3F), -121,2 (m, 1F), 131,4 (m, 1F), -134,4 (m, 1F), -165,1 (m, 1F).
1H NMR (, ppm): 4,2 (s, 2H).
2,3,5,6-Tetrafluoro-4(trifluoromethyl)benzyl chloride (1e)
2,3,5,6-Tetrafluoro-4(trifluoromethyl)benzyl chloride was obtained similar to (1f) in 7% yield. B.p. 96-98°С /20 mm Hg.
19F NMR (, ppm):-143,3 (m, 4F), -58,0 (m, 3F).
1H NMR (, ppm): 4,1 (s, 2H).
3,4-difluorobenzyl chloride
To a solution of 1,2-difluorobenzene (270 g, 2,37 mol) in chloroform (540 ml) concentrated sulfuric acid (580 ml) was gradually added with stirring. The solution was cooled to a temperature of 5°C and freshly prepared bis(chloromethyl) ether (273 g , 2,37 mol) was added dropwise with vigorous stirring over 1,5 hours, maintaining the temperature in the range of 3-7°C. After adding bis (chloromethyl) ether, the reaction mixture was stirred for 1 hour, maintaining the temperature at the same range. Then the mixture was allowed to warm to room temperature, and poured onto crushed ice. The organic layer was separated, washed with water until neutral, dried with CaCl2 and a main part of the solvent was distilled off, the residue was distilled in vacuo, collecting a fraction boiling 80°С/15 mm Hg. Low boiling fraction was collected in a trap, cooled with dry ice-acetone. Further distillation of the trap content yielded 1,2-difluorobenzene (70 g). Distillation of the main fraction gave 3,4-difluorobenzyl chloride (168 g), b.p. 80°С/15 mm Hg, yield 58% (taking into account 74% conversion).
19F NMR (, ppm):-136,3 (m, 1F), -135,2 (m, 1F).
1H NMR (, ppm): 4,3 (s, 2H), 6,8-7,2 (m, 3H).
1,2-bis(chloromethyl)-4,5-difluorobenzene
1,2-bis(chloromethyl)-4,5-difluorobenzene was obtained by adding bis(chloromethyl) ether to 1,2-difluorobenzene in sulfuric acid without solvent under cooling.
19F NMR (, ppm): -136,2 (m, 2F).
1H NMR (, ppm): 4,22 (s, 4H), 6,75 (t, 2H).
Acknowledgments
This work was performed with the financial support from Ministry of Science and Higher Education of the Russian Federation using the equipment of Center for molecular composition studies of INEOS RAS.
References
- L.I. Belen'kii, Yu.B. Vol'kenshtein, and I.B. Karmanova, Russ.Chem. Rev., 1977, 46(9) 891-903, translated from Uspekhi Khimii, 1977, 46, 1698-1719.
- C. Fuson, C.H, McKeever, "Chloromethylation of Aromatic Compounds", Organic Reactions, American Cancer Society, 2011, 63–90.
- A.A. Shtark, V.D. Shteingarts, Izvestiya Sibirskogo Otdeleniya Akademii Nauk SSSR, Seriya Khimicheskikh Nauk, 1976, 4, 123-127 (in Russian).
- CN101973850, 2011.
- DE1221234, 1966.
- DE1226116, 1966.
- GB1065041, 1967.
- M. Camps, M. Chatzopoulos, J.-M. Camps and J.-P. Montheard, J. Macromol. Sci. Part C: Polym. Rev. 1987, 27, 505.
- H. Zhang and Z. Zhou, J. Appl. Polym. Sci. 2008, 110, 1756.
- C. Yan, S. Zhang, D. Yang and X. Jian, J. Appl. Polym. Sci. 2008, 107, 1809.
- CN108530301, 2018.
- R. Baltzly , L. W. Sheehan, A. Stone, J. Org. Chem. 1961, 26, 7, 2353-2355.
- CN109761743, 2019.
- a). Sintezy ftororganicheskikh soyedineniy, red. Igumnov S.M.. Igumnova E.V. Moscow, 2011,
tom 2 (in Russian).
b). Sintezy ftororganicheskikh soyedineniy, red. Igumnov S.M.. Igumnova E.V Moscow, 2015, tom 2 (in Russian).
c). Sintezy ftororganicheskikh soyedineniy, red. Igumnov S.M.. Igumnova E.V. Moscow, 2018, tom 2 (in Russian). - "Organic syntheses", Vol. 36, 1956, p.1,
ARTICLE INFO
Received 02 December 2019
Accepted 17 December 2019
Available online December 2019
Recommended for publication by Prof. V. Kornilov
Fluorine Notes, 2019, 127, 7-8
