Received: June 2019
DOI 10.17677/fn20714807.2019.04.01
Fluorine Notes, 2019, 125, 1-2
PREPARATION OF PENTAFLUOROPHENOL AND OTHER POLYFLUOROPHENOLS AND POLYFLUORODIHYDROXYBENZENES FROM POLYFLUOROAROMATIC ACIDS
V.E. Boyko, V.L. Don, S. M. Igumnov
A.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilov St. 28, Moscow, 119991, Russian Federation
e-mail: boykii@mail.ru
NPO PIM-INVEST LLC, Leninsky Prospect, 47, Moscow, 119991, Russian Federation
Abstract: A process developed for the preparation of pentafluorophenol and other polyfluorophenols and polyfluorodihydroxybenzenes from corresponding aromatic acids by oxidation of organozinc compounds, obtained by decarboxylation of trimethylsilyl esters of these acids. The reactions of preparing zinc-organic compounds and their oxidation were carried out without isolation of intermediates.
Keywords: pentafluorophenol, pentafluorobenzoic acid, oxidation, polyfluorophenol, polyfluordihydroxibenzene, perfluorobenzoic acid (trimethyl)silyl ester.
Fluorinated phenols are widely used for producing polymers possessing unique properties [1] and as components of metallocene catalyst systems for olefin polymerization [2-4].
Pentafluorophenyl esters's formation is broadly applied in peptide synthesis since pentafluorophenol readily reacts with amines to form amide bonds, thus, pentafluorophenol is used in the synthesis of peptides and nucleosides, that are intermediates in the synthesis of antitumor agents, inhibitors of HIV [5-7].
Most of known methods for preparing pentafluorophenol involves substituting of fluorine atom in hexafluorobenzene for OH-group by reacting it with alkalis, or preparing phenyl-alkyl ethers by reacting hexafluorobenzene with metal alcoholates and their further decomposition to pentafluorophenol.
A major obstacle to the industrial use of these methods is that hexafluorobenzene is not a commercially available product now in connection with that hexachlorobenzene from which it has been previously obtained now is prohibited for production due to its high carcinogenicity. It is therefore an urgent need for a process for synthesizing pentafluorophenol from commercially available raw material, namely, pentafluorobenzoic acid, pentafluorobenzene or bromopentafluorobenzene.
Preparation of pentafluorophenol from bromopentafluorobenzene via pentafluorophenyl magnesium bromide followed by its conversion to pentafluorophenyldimethoxyborate or pentafluorophenylboronic acid by reaction with trimethoxyboron, and further oxidation by hydrogen peroxide [8 -9], or by direct oxidation of pentafluorophenyl magnesium bromide with peroxides [10] is described. However, the preparation of the Grignard reagent in diethyl ether or tetrahydrofuran is a high hazard process and requires the use of an inert atmosphere.
Pentafluorobenzoic acid and other polyfluoroaromatic acids are currently the most available commercial fluoroaromatics, therefore, it has been appropriate to find a way to polyfluorphenols starting from polyfluoroaromatic acids. We have developed previously convenient methods for production of pentafluorophenyl (trimethyl)silane from pentafluorobenzoic acid (decarboxylation of aromatic acid's salts in the presence of silylating agents or decarboxylation of corresponding trimethylsilyl ethers) [11,12]. Then we investigated production of pentafluorophenol by oxidation of trimethyl(pentafluorophenyl)silane and found that an oxidation of the silane with t-butylperoxybenzoate in the presence of KF and CuCl in DMF gave pentafluorophenol with 65% yield, but the reaction was complicated by the formation of by-products - pentafluorobenzene and decafluorobiphenyl. When hydrogen peroxide and t-butyl peroxide were used as oxidizing agents pentafluorobenzene was the main product of the reaction, and no pentafluorophenol formation occurred at all [13].
Further searches of the approaches to the preparation of pentafluorophenol and other polyfluorobenzohydroxybenzenes from corresponding aromatic acids have led us to investigation of an oxidation of zinc-polyfluoroaromatic compounds. The advantage of zinc-organic compounds in comparison with magnesium-organic compounds is their greater stability, which makes it possible to work with them not in ethers but in polar aprotic solvents such as DMF.
A solution of the zinc-organic compound was prepared from pentafluobenzoic acid in two steps without isolation of an intermediate product from the trimethylsilyl ester of the acid. Pentafluorobenzoic acid was refluxed with an excess of trimethylchlorosilane, and the resulting trimethylsilyl ether was then converted to the corresponding zinc derivative without isolation by reaction with zinc salts or zinc oxide dehydrated previously by azeotropic distillation of water.
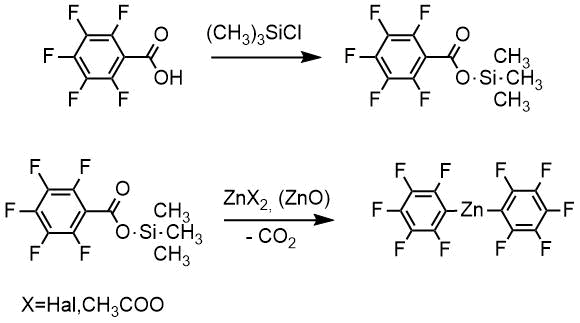
The zinc-organic compound solution thus obtained was oxidized with tert-butylperoxybenzoate in the presence of cuprous salts CuX, where X = Cl, Br, I (method A ) to give pentafluorophenol tert-butyl ether. Further refluxing of tert-butyl ether of pentafluorophenol with hydrochloric acid gave pentafluorophenol. A total yield of the product (99% pure) was about 70%.
method A
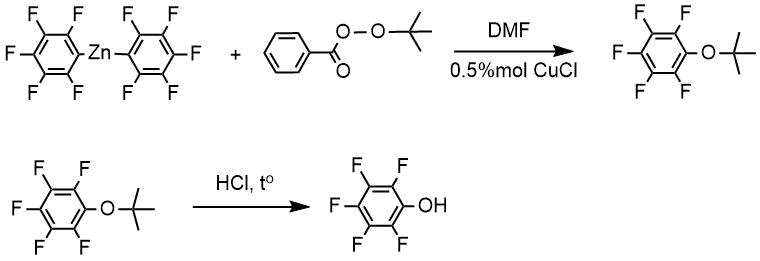
If urea peroxide was taken as a peroxide compound (method B), pentafluorophenol was obtained at once as a result of the reaction of urea peroxide with the corresponding zinc derivative with further treatment of the reaction mixture with hydrochloric acid, however, the yield of pentafluorophenol in this case was significantly less because pentafluorobenzene was obtained along with pentafluorophenol.
method B
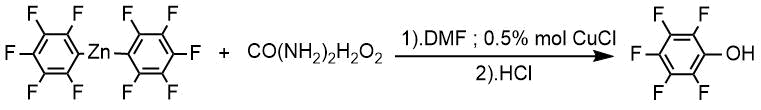
According to the method A 2,4,5,6-tetrafluorophenol was obtained from 2,4,5,6-tetrafluorobenzoic acid with 76% yield.
If dicarboxylic perfluoroaromatic acid was used as a starting material, bis-(trimethyl)silyl ester of corresponding acid was obtained, and then according to the method A through intermediate formation of bis-zinc derivative solution, corresponding bis-(tert-butyl) ether followed by dihydroxytetrafluorobenzene was obtained.
From tetrafluoroterephthalic acid in reaction with an excess of trimethylchlorosilane, bis-trimethylsilyl ether of tetrafluorterephthalic acid was obtained, and further by the method A-tetrafluorohydroquinone 99% purity in 80% yield.
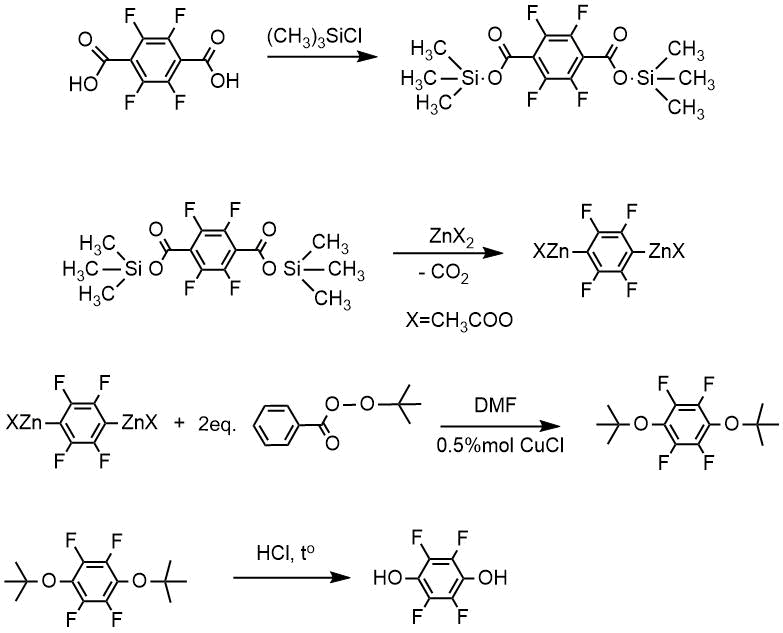
Thus, the proposed method allows to produce hydroxy compounds from corresponding fluorinated aromatic acids via an intermediate formation of zinc-organic compounds without isolation of the intermediates. The process for preparing polyfluorohydroxybenzenes is provided by the Russian Federation Patent No 2536872.
Experimental Part
NMR 1H, 19F spectra were recorded on a “Bruker AVANCE-300” spectrometer at 300 and 282 MHz, respectively, exterior standard CDCl3. Chemical shifts for 1H spectra were determined relatively to proton signal of solvent (CDCl3) and given in ppm relative to TMS Chemical shifts of 19F spectra were determined in ppm relatively to CFCl3. Downfield shift are positive.
Pentafluorobenzoic acid trimethylsilyl ester
Pentafluorobenzoic acid (1493 g, 7.04 mol) was added portionwise to trimethylchlorosilane (2300 g, 21.18 mol) under stirring. The reaction mixture was heated to reflux and refluxed until the gas evolution was complete. Then the excess of trimethylchlorosilane was distilled off to the temperature of the residue 120 -130°C and thus 2000 g of the pentafluorobenzoic acid (trimethyl)silyl ester was obtained, which was used further without purification. The yield was quantitative.
NMR 19F δ, ppm.: -140 m.(2F), -152 m.(1F), -163 m. (2F). 1H NMR δ, ppm.: 0,78 s.
tert-Butoxypentafluorobenzene
A mixture of toluene (800 g), DMF (1200 g) and ZnAc22H2O (430 g, 1,51 mol) was heated to reflux and water was separated with a Dean-Stark trap. After completion of water separation, the Dean-Stark trap and the reflux condenser were replaced with a dropping funnel and a distillation system, consisting of a fractionating column, Liebig condenser, a receiver adaptor and a flask, pentafluorobenzoic acid trimethylsilyl ester (1000 g, 3,52 mol) was added dropwise, then the reaction mixture was heated to 140-150°C and in the same time fraction with the boiling point 120-130°С was distilled off. CuCl (2 g, 0,02 mol) was added with stirring, and then tert-butylperoxybenzoate (760 g, 3,91 mol) was added dropwise at 20-25°С. The reaction mixture was stirred for 4 hr, then cooled to 10-15°С and diluted with HCl (2l, 10% solution), then stirred for 20 minute.The upper layer was decanted, 2-2,5 volume of water was added to the lower layer and it was distilled with water.
A crude product (783 g) was obtained, containing 88% of tert-butoxypentafluorobenzene (by GC), it was used further without purification.
NMR 19F δ, ppm.: -153,5 m.(2F), -165,0 m.(1F), -166,8 m. (2F). 1H NMR δ, ppm.: 0,78 s.
Pentafluorophenol (method A)
To the crude tert-butoxypentafluorobenzene (783 g) two volumes of 35% HCl solution was added, the resulting mixture was heated to reflux and refluxed for 4 hr (until the completion of gas evolution). Then the reaction mixture was cooled to 10°C, the aqueous layer was decanted and the organic (lower) was rectified and 475 g of a pentafluorophenol (99% pure by GC) was isolated. B.p. 139-142°С, M.p. 34-36°С. The yield 72%.
NMR 19F δ, ppm.: -165,5 m.(2F), -168,6 m.(2F), -174,6 m. (1F).
Pentafluorophenol (method B)
A mixture of toluene (800 g), DMF (1200 g) and ZnO (122g, 1,51 mol) was heated to reflux and water was separated with a Dean-Stark trap. After completion of water separation, the Dean-Stark trap and the reflux condenser were replaced with a dropping funnel and a distillation system, consisting of a fractionating column, Liebig condenser, a receiver adaptor and a flask. Pentafluorobenzoic acid trimethyl silyl ether (1000 g (3.52 mol)) was added dropwise to the reaction mixture, then the residue was heated to a temperature of 140 -150°C, while distilling off a fraction with bp 120 -130°C.
Reaction mixture was cooled to 10-15°С, CuCl (2 g, 0,02 mol) was added with stirring, and then urea peroxide (500 g) was added in portions and reaction mass was stirred for 1 -2 h. The content of the flask are then poured into hydrochloric acid (2.5l of 10% solution), the mixture was stirred for 10 minutes, the lower layer was separated and steam distilled over concentrated hydrochloric acid. The crude product obtained was rectified and pentafluorophenol (360 g of 99% purity) and 267 g of a pre-run, containing pentafluorobenzene were obtained.
2,3,5,6-Tetafluorobenzoic acid trimethylsilyl ester
Tetrafluorobenzoic acid (120 g, 0,62 mol) was added with stirring to trimethylchlorosilane (202 g, 1,86 mol). The reaction mixture was heated to reflux to a completion of gas evaluation, then an excess of trimethylchlorosilane was distilled off while heating the residue to a temperature 120-130°С.
2,3,5,6-Tetafluorobenzoic acid trimethylsilyl ester (165 g) obtained was used further without additional purification.
NMR 19F δ, ppm.: 139,8 m. (2F), -140,6 m. (2F), 1H NMR δ, ppm.: 0,78 s.
2,3,5,6-Tetrafluorophenol (method A)
Toluene (200 g) and zinc acetate dihydrate (68 g) were placed in a flask, the reaction mixture is heated to reflux (110 -115°С) and water was distilled off. After the separation of water had completed, the reaction mixture was cooled and the water was weighed out. If all of the required amount of water had been released, then DMF (300 g) was added to the reaction mixture, the reaction mixture was heated again to reflux and toluene was distilled off to 130°С in vapors. The reaction mixture was cooled and 2,3,5,6-tetrafluorobenzoic acid trimethylsilyl ester (165 g, 0,62 mol) was added dropwise with stirring. Self-heating of the reaction mixture and dissolution of zinc acetate were observed. The reaction mixture was then heated, and at a temperature of 80 - 85°С, gas evolution begun and (trimethyl)silyl acetate begun to distill off. The distillation was continued to a residue temperature of 140 -150°C.
To the solution of Zn-derivative of 2,4,5,6-tetrafluorobenzene thus obtained, CuCl (0,6 g) was added with stirring and then tert-butylperoxybenzoate (0,62 mol) was added dropwise at 20-25°С and the reaction mixture was stirred for 1 hr. The content of the flask was then poured into hydrochloric acid (0,5l of 10% solution), the mixture was stirred for 10 minutes, the lower layer was separated and refluxed with concentrated hydrochloric acid for 2 hr, and then again separated and steam distilled over concentrated hydrochloric acid to obtain 2,4,5,6-tetrafluorophenol of 99% purity.(The yield 76%)
NMR 19F δ, ppm.: 141,7m. (2F), -161,7m. (2F).
Tetrafluoroterephthalic acid bis (trimethylsilyl) ester
Trimethylchlorosilane (182 g, 1,69 mol) was added to tetrafluoroterephtalic acid (50 g, 0,21 mol), the reaction mixture was refluxed to the completion of the gas evolution, then an excess of trimethylchlorosilane was distilled off. Tetrafluoroterephthalic acid bis-(trimethyl)silyl ester (80,2 g, 0,21 mol) was obtained.
19F NMR(CDCl3), δ, ppm.:-141м (4F), 1H NMR(CDCl3) δ, ppm: 0,78c.
Tetrafluorohydrohinone
DMF (250 ml) and anhydrous zinc acetate (38,5 g) were added to tetrafluoroterephthalic acid bis (trimethylsilyl) ester (80,2 g, 0,21 mol) and the reaction mixture was heated while distilling off trimethylsilyl acetate and decarboxilation. After the completion of the gas evolution the reaction mixture was cooled to 0°С, CuCl (0,2 g) was added and tert-butylperoxybenzoate (81 g , 0,42 mol) was added dropwise. The reaction mixture was stirred for additional 2 hr at 0 оС and 6 hr at ambient temperature, poured on ice mixed with 10% HCl solution, a lower layer was separated. Two volumes of hydrochloric acid were added to the lower layer, the resulting mixture was refluxed for 3 hr, and then cooled, the water (upper) layer was decanted, the lower layer was distilled to yield tetrafluorohydrohinone (30 g) of 99% purity. The yield 80%.
NMR 19F δ, ppm.: 165,3m (4F).
Acknowledgments
This work was performed with the financial support from Ministry of Science and Higher Education of the Russian Federation using the equipment of Center for molecular composition studies of INEOS RAS.
Literature
- B. Boutevin, A. Rousseau, D. Bosc, J. Polym. Sci. A, Polym.Chem., 1992, vоl. 30(7), p.1279-1286.
- Patent US8501655, 2013.
- Franz A.R. Kaul, Gerd T. Puchta, Horst Schneider, Manja Grosche, Dimitrios Mihalios, Wolfgang A. Herrmann J. Organomet. Chem., 2001, vol. 621, p.184.
- Metz, Matthew V.; Sun, Yimin; Stern, Charlotte L.; Marks, Tobin J. Organometallics, 2002, 21(18), p.3691-3702.
- Patent US7262169, 2007.
- Patent Application WO9616931, 1996.
- Jérôme Grugier, Juan Xie, Isabelle Duarte, Jean-Marc Valéry J. Org. Chem., 2000, vol. 65(4), p. 979.
- Korenaga T., Kobayashi F., Nomura K., Nagao S., Sakai T. J. Fluorine Chem., 128, 2007, p. 1153-1157.
- Patent JP 82548, 2005.
- Patent CN1847210, 2006.
- Patent RU2507209.
- Patent RU2521168.
- Boiko V.E., Tyutyunov A.A., Don V.L., Igumnov S.M., Fluorine Notes, 2013, 91, p. 9-10.
ARTICLE INFO
Received 13 June 2019
Accepted 15 July 2019
Available online August 2019
Recommended for publication by PhD Andrey A. Tyutyunov
Fluorine Notes, 2019, 125, 1-2
