Received: March 2018
DOI 10.17677/fn20714807.2018.02.01
Fluorine Notes, 2018, 117, 1-2
Reactions of Perfluoroaliphatic Sulphonyl Halides with Phosphorus Trihalides
A.A. Tyutyunovab, L.F. Ibragimovaa, N.D. Kagramanova, S.R. Sterlina, S.M. Igumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, V-334, GSP-1, Moscow, 119991 Russia
bNPO PiM-INVEST LLC, ul. Vavilova 28, Moscow, 119991 Russia
e-mail: tuytuynov@rambler.ru
Abstract: It is shown that fluoroaliphatic sulfonyl halides are reduced under the action of phosphorus trihalides with formation of the corresponding disulfides, including those containing such functional groups as fluorosulfonyl, carboxymethyl, and also trifluorovinyloxy groups. Reaction intermediates are perfluoroalkane sulfinyl halides and perfluoroalkane sulfene halides. Formation of the latter is confirmed by synthesis of adducts of the corresponding sulfene halides with unsaturated hydrocarbons.
Key words: trifluoromethane sulfonyl bromide, perfluoroalkane sulfonyl halides, diperfluoroalkyl sulfides, phosphorus trichloride, phosphorus tribromide.
Deoxygenation of S–O derivatives in a reaction with phosphorus halides was first described in 1871 by Michaelis [1] and was later studied by a number of authors [2-4]. It was later shown that this reaction was of a general character and could be used for reduction of aliphatic and aromatic sulfonyl halides, sulfi- and sulfoacids or their salts to the corresponding disulfides [5-9]; here, it was found that the reducing potential of PBr3 considerably exceeded the reducing potential of PCl3. In their turn, sulfonyl bromides underwent reduction much more easily than sulfonyl chlorides. At the same time, it was demonstrated that the reaction of benzene sulfonyl chloride with PBr3 at t>150°C resulted in formation of PhSO2Br in >90% yield but not the products of its reduction [10].
The aim of this work is to elucidate the regularities of interaction between perfluoroaliphatic derivatives of sulfi- and sulfoacids with phosphorus trihalides.
We have found that interaction of trifluoromethane sulfonyl bromide (1) with phosphorus trichloride (at the molar ratio of 1 : PCl3 = 1:1) proceeds already at the room temperature with the formation of trifluoromethane sulfinyl chloride (2) that can be isolated from the reaction mixture in 40% yield [11].
Scheme 1
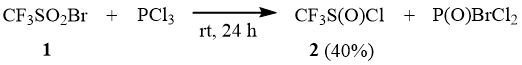
Here, the reaction also results in formation of P(O)Cl3, bromine, and products of deeper reduction of 1.
The reaction of 1 with two equivalents of PCl3 at room temperature in the presence of catalytic amounts of DMF results in formation of disulfide 3 and bromine. The reaction of CF3SO2Br with PBr3 occurs in a similar way. In this case, the reaction does not require catalysis by DMF, but leads to formation of considerable amounts of P(O)Br3 as precipitate (m.p. 50°C). Therefore, it is more convenient to carry it out in the medium of acetonitrile, as the reaction accelerates in the presence of acetonitrile and phosphorus oxybromide does not precipitate. At molar ratio of CF3SO2Br : PBr3 = 1:1, conversion of 1 is 50% and formation of disulfide 3 is observed. This is probably related to instability of the initially formed CF3S(O)Br that, as shown earlier, easily disproportionate with formation of CF3SO2Br and relatively unstable CF3SBr [12]. Meanwhile, in the case of the ratio of CF3SO2Br : PBr3 = 1:2, all reacting sulfonyl bromide 1 is reduced to disulfide 3.
Scheme 2
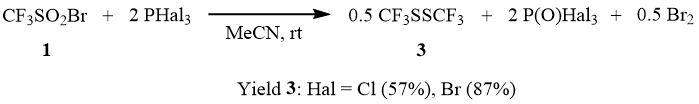
We found that trifluoromethane sulfinyl chloride (2) does not react with PCl3 even under heating to 90÷100°C. In the presence of catalytic amounts of DMF, 2 reacts slowly with PCl3 already at the room temperature. However, even after the heating of the reaction mixture in a sealed ampoule at 90÷100°C for 5–6 h, conversion of 2 is only 58%; and a mixture of disulfide 3 and sulfene chloride 4 is formed.
Scheme 3
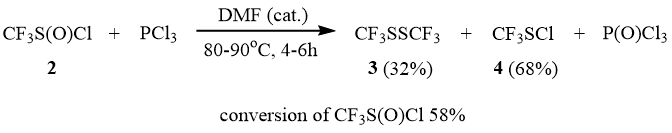
As distinguished from 1, sulfinyl chloride 2 reacts slowly with PBr3 only at 90÷100°C. However, in the presence of catalytic amounts of DMF, the reaction occurs slowly already at the room temperature. Here, in addition to disulfide 3 as the main product, the reaction yields a side product, sulfonyl chloride 5, which is most probably related to disproportionation of the intermediate trifluoromethane sulfinyl bromide (see above). The further substitution of the sulfonyl bromine atom for chlorine results in formation of 5 that is not reduced by PBr3 under these conditions.
Scheme 4
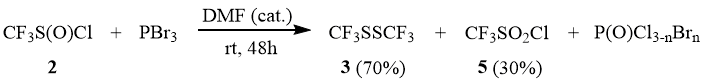
The reaction of trifluoromethane sulfonyl chloride (5) with PBr3 in the acetonitrile medium occurs at 100°C in a sealed ampoule for 3 h with formation of disulfide 3. Sulfonyl chloride 5 does not react under these conditions with the less reactive PCl3.
Scheme 5
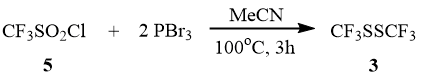
Unlike trifluoromethane sulfonyl bromide (1) and chloride 5, trifluoromethane sulfonyl fluoride does not react with PBr3 in the acetonitrile medium in a sealed ampoule at 100°C, which is most probably due to the greater strength of the S–F bond (~343 kJ/mol) as compared to the S–Cl and S–Br bonds (~241 and ~218 kJ/mol, respectively) [13]. The result obtained agrees to a certain extent with the resistance of thionyl fluoride to PCl3 exposure [14].
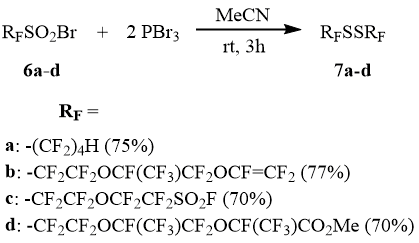
Higher perfluoroalkane sulfonyl bromides exposed to PBr3 in the acetonitrile medium are also smoothly converted to the corresponding disulfides containing such functional groups as the fluorosulfonyl, carboxymethyl, and also trifluorovinyloxy groups.
Scheme 6
In contrast to 6b, the reaction of vinyloxysulfonyl bromide 8 with PBr3 results in formation of cyclic oxathiolane 9 that is most probably the product of intramolecular addition of the intermediate sulfene bromide to the trifluorovinyloxy group. One must point out that using PCl3 in this reaction leads to formation of a mixture of BrCF2-oxathiolane 9 and ClCF2-oxathiolane 9’ at the ratio of 3:1.
Scheme 7
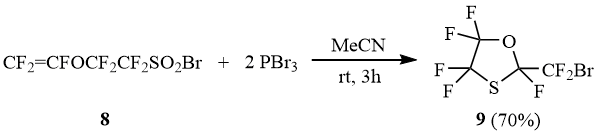
Such cyclisation’s resulting in formation of five–membered cycles are specific for perfluoroaliphatic compounds in general. In the literature, there are examples of nucleophilic and free radical formation of perfluorinated 1,3-oxathiolane-3,3-dioxides [15, 16], as well as a number of other reactions resulting in formation of five–membered cyclic products [17, 18].
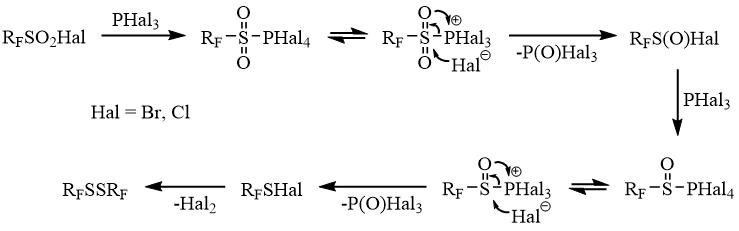
In all probability, reduction of sulfonyl and sulfinyl halides occurs stepwise with participation of sulfonyl and sulfinyl tetrahalogen phosphoranes as primary reaction intermediates.
Scheme 8
Generation of fluoroaliphatic sulfene halides as a result of reduction of fluoroalkane sulfonyl halides by phosphorus trihalides allows assuming that in the presence of unsaturated hydrocarbons this reaction can lead to formation of vicinal fluoroalkylthio halogenides (electrophilic addition of CF3SCl to cyclic and terminal olefins was described earlier [19]).
Indeed, the reaction of 1 with PCl3 in the presence of cyclohexene results in formation of trifluoromethylthio-β-halogen cyclohexanes 10-11 containing both chlorine and bromine atoms.
Scheme 9
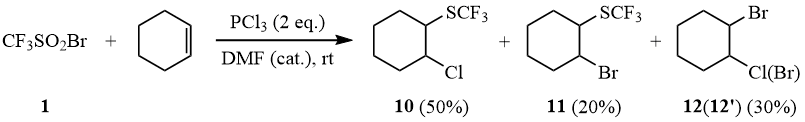
In its turn, reduction of sulfonyl bromide 1 by PBr3 in the presence of cyclohexene in acetonitrile solution results in formation of a mixture of products: 11 (84%), 12’ (16%).
Similarly, the reaction of ethyl cyclopent-3-enyl carboxylate with 1 and PCl3 results in formation of a mixture of stereoisomers of trifluoromethylthio chlorides and bromides.
In the case of terminal olefins that are less active than cycloalkenes, this reaction occurs either predominantly with formation of a mixture of Markovnikov (M) and anti-Markovnikov (a-M) products of addition of sulfene halides to olefins or with formation of disulfide 3 and alkene dibromide.
Thus, reaction of 1 with PCl3 in the presence of allyl trifluoroacetamide leads to formation of a mixture of regioisomers 13-14 and a negligible amount of disulfide 3 and N-2,3-dibromopropyl trifluoroacetamide.
Scheme 10
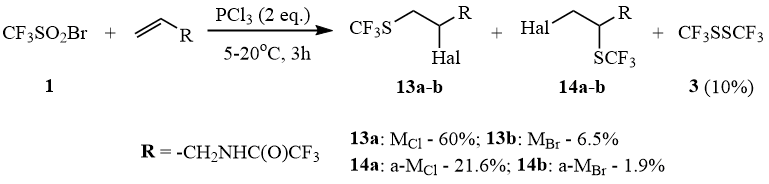
A similar reaction with PBr3 results in formation of only disulfide 3 as the only organofluorine compound and allyl trifluoroacetamide dibromide, which can be explained by the relatively low stability of trifluoromethane sulfene bromide.
Essentially the same result was obtained in the reaction of CF3SO2Br with PCl3 in the presence of allyl bromide.
Scheme 11
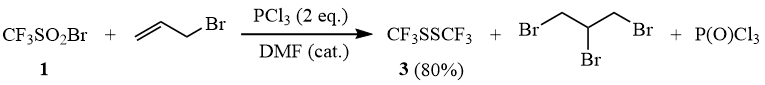
When PCl3 reacts with sulfonyl bromide 1 in the presence of allyl acetate and catalytic amounts of DMF, both reaction versions are realized: a mixture of regioisomers of the both products of conjugated halogen trifluoromethylthiolation of allyl acetate alongside with disulfide 3 and 2,3-dibromopropyl acetate are formed in commensurable amounts (3 – 28%, MCl – 42%, a-MCl – 11%, MBr – 12%, a-MBr – 7%).
Sodium trifluoromethane sulfinate (15) reacts with PBr3 with formation of disulfide 3 (similarly to formation of diphenyl sulfide in the reaction of PCl3 with PhSO2Na [5]). Most probably, formation of 3 is preceded by reduction of 15 up to trifluoromethane sulfinyl bromide (see [20]) that is further transformed into 3 (cf. scheme 8).
Scheme 12
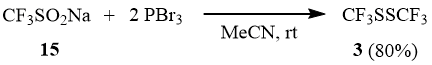
Registration of products of stepwise reduction of fluoroalkane sulfonyl halides by phosphorus trihalides (RFSO2Hal→RFSOHal→RFSHal→RFSSRF) allows assuming that the chain of transformations in Scheme 8 is unitary. In this connection, one should point out that the appearance of trifluoromethylthio derivatives obtained by the reaction of electron–donating heteroaromatic compounds with a mixture of CF3SO2Na and PCl3 [21] can be explained not only by reduction of the corresponding trifluoromethyl sulfoxides but also by the interaction of the starting compounds with the intermediate trifluoromethane sulfene chloride 4 [22].
It is thus shown that reduction of perfluoroalkane sulfonyl bromides with phosphorus trihalides can be used for synthesis of fluoroaliphatic disulfides, including functionalized ones and, in some cases, products of perfluoroalkyl thiolation of unsaturated compounds.
Experimental
1H, 19F, and 31P NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300, 282, and 121 MHz, accordingly; the external standard was CDCl3. Chemical shifts for 1H spectra are presented vs. the residual signal of the solvent (δ 7.26) and are given in ppm vs. tetramethylsilane. Chemical shifts in 19F spectra are given in ppm vs. CFCl3. Downfield shifts are positive. Chemical shifts in 31P spectra are given vs. 85% H3PO4. Mass spectra are recorded using a Finnigan Polaris Q mass spectrometer (Trace GC ultra). Elemental analysis was carried out in Laboratory of Microanalysis of A.N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences. The method of synthesis of sulfonyl bromides 1, 6c-d, and 8 was described earlier [16, 23-24]. Sulfonyl bromide 6a was obtained in a similar way. Sulfonyl bromide 6b was obtained in 60% yield from the corresponding sulfonyl fluoride by its reduction to sodium sulfinate by exposure to Na2SO3 with the further bromination.
Reaction of CF3SO2Br with 1 equivalent of PCl3.
A mixture of 102.76 g (0.482 mol) of trifluoromethane sulfonyl bromide (1) and 66.25 g (0.482 mol) of phosphorus trichloride is conditioned for 20–24 h at 20÷25°C, then boils to boil under reflux on Vigreux column distilling off low boiling products (25÷60°C) to a cooled receiver (0÷5°C); 62.4 g of distillate are collected. According to 19F-NMR data it contains 5.8% CF3SSCF3 (δ: –47); 5.8% CF3SCl (δ: –51); 15% CF3SO2Br (δ: –78), and 73% CF3S(O)Cl (δ: –79). According to 31P-NMR data the distillation residue is a mixture of P(O)BrCl2 (δ: –29) and P(O)Cl3 (δ: 2) in the molar ratio of 1:1.
To the distillate obtained 5 g of cyclohexene is added at 0÷5°C and sulfinyl chloride 2 (b.p. 42÷44°C) is isolated by rectification (30 g; 40%); 19F NMR δ: –77 (s, CF3).
The obtained product was used in the further syntheses without additional purification.
Reduction of sulfonyl bromides 1, 6a-d, 8 by phosphorus tribromide (general procedure).
0.1 mol of sulfonyl bromide (1, 6a-d or 8) was added dropwise to the mixture of 54.14 g (0.2 mol) of phosphorus tribromide and 30 ml of acetonitrile under stirring at 5÷10°C. The mixture was stirred at 20÷25°C for 3 h. Then the reaction mixture was poured portionwise onto a crashed ice; the lower organic layer was separated, washed several times with water and distilled over P2O5.
The obtained product is additionally purified by rectification.
Hexafluorodimethyl disulfide (3).
Yield 87%; b.p. 33÷34°C. 19F NMR δ: –48 (s, SCF3) (before distillation, traces of Br2 are removed by washing the product with aqueous Na2SO3).
Disulfide 3 can also be obtained by the reaction of sulfonyl bromide 1 with PCl3 in 57% yield.
Di-4-hydroperfluorobutyl disulfide (7a).
Yield 75%; b.p. 189°C. Found (%): C, 19.28; H, 0.44; F, 64.77; S, 13.64. C8H2F16S2. Calculated (%): C, 20.61; H, 0.43; F, 65.20; S, 13.75. 1H NMR δ: 5.87 (tt, 1H, 2JHF = 52 Hz, 3JHF = 4.8 Hz, HCF2); 19F NMR δ: –139.1 (d, 4F, 2JFH = 52 Hz, HCF2), –130.6 (s, 4F, CF2), –122.4 (s, 4F, CF2), –91.1 (s, 4F, CF2S).
Di(perfluoro-3,6-dioxa-4-methyl octene-7-yl) disulfide (7b).
Yield 77%; b.p. 71÷73°C/0.5 Torr. Found (%): C, 21.30; F, 62.58; S, 8.04. C14F26O4S2. Calculated (%): C, 21.28; F, 62.51; S, 8.11. 19F NMR δ: –147.3 (t, 1F, 3JFF = 21 Hz, CFCF3), –138,7 (dd, 1F, 3JFF-trans = 113 Hz, 3JFF-cis = 68 Hz, OCF=CF2), –125,2 (dd, 1F, 2JFF = 85 Hz, OCF=CF2-trans), –117.6 (dd, 1F, 2JFF = 85 Hz, OCF=CF2-cis), –96.8 (s, 2F, CF2S), –87,3 (br. s, 2F, CF2O), –84.5 (m, 2F, CF2O), –82.7 (d, 3F, 3JFF = 6 Hz, CF3).
Di-(5-fluorosulfonyl perfluoro-3-oxaamyl) disulfide (7c).
Yield 70%; b.p. 54÷55°C/0.3 Torr. Found (%): C, 14.29; F, 51.51; S, 19.38. C8F18O6S4. Calculated (%): C, 14.51; F, 51.63; S, 19.36. 19F NMR δ: –114 (s, 4F, CF2SO2F), –96 (s, 4F, CF2S), –85.8 + –83.6 (two s, 8F, CF2OCF2), 43.9 (s, 2F, SO2F).
Di-(7-methoxycarbonyl perfluoro-3,6-dioxa-4,7-dimethylheptyl)disulfide (7d).
A mixture of stereoisomers. Yield 70%; b.p. 112÷122°C/0.3 Torr. Found (%): C, 22.75; H, 0.65; B, 56.35; F, 6.59. C18H6F28O8S2. Calculated (%): C, 22.85; H, 0.64; F, 56.21; S, 6.78. 1H NMR δ: 3.97 (s, 3H, CH3); 19F NMR δ: –146.5 (dd, 2F, CF2OCFCF2O), –133,8 (dd, 2F, OCFCO2Me), –98.5 (br. s., 4F, CF2S), –90.6÷–89.5+–84.3÷–83.2 (m, 4F, OCFCF2O), –87.7 (s, 6F, CF3), –87.5÷–86.5 (m, 4F, SCF2CF2O), –85.2 (s, 6F, CF3).
2-(Bromodifluoromethyl)-2,4,4,5,5-pentafluoro-1,3-oxathiolane (9).
Yield 70% (a mixture of enantiomers); b.p. 94÷95°C. Found (%): C, 15.50; Br, 26.10; F, 42.91; S, 10.05. C4BrF7OS. Calculated (%): C, 15.55; Br, 25.86; F, 43.04; S, 10.38. 19F NMR δ: –93.4, –87.2 (ABq, 2F, 2JFF = 209 Hz, CF2S), –92.8, –83.3 (ABq, 2F, 2JFF = 131 Hz, CF2O), –86.6 (s, 1F, CF), –67.3 (s, 2F, CF2Br). The mass spectrum (M/Z, reference): 308[M]+, 289[M–F]+, 261[M–COF]+, 229[M–Br]+, 201[M–Br–C–O]+, 179[M–CF2Br]+(100%), 129[CF2Br]+, 119[C2F5]+, 113[C2F3S]+, 100[C2F4]+, 82[CF2S]+, 69[CF3]+, 63[CFS]+, 47[COF]+.
2-(Chlorodifluoromethyl)-2,4,4,5,5-pentafluoro-1,3-oxathiolane (9').
Obtained in a mixture with bromide 9 at the ratio of 9/9'=3/1 in the reaction of 8 with two equivalents of PCl3.
The mass spectrum (M/Z, reference): 245[M–F]+, 229[M–Cl]+, 217[M–COF]+, 201[M–Cl–C–O]+, 179[M–CF2Cl]+(100%), 163[C2F4SCF]+, 135[C2F4Cl]+, 119[C2F5]+, 113[C2F3S]+, 100[C2F4]+, 85[CF2Cl]+, 82[CF2S]+, 69[CF3]+, 63[CFS]+, 47[COF]+.
Reaction of trifluoromethane sulfonyl bromide 1 with PCl3 (2 eq.) in the presence of olefins (general procedure).
1 drop of dimethylformamide is added to a mixture of 27.5 g (0.2 mol) of phosphorus trichloride and 0.1 mol of the olefin, then of sulfonyl bromide 1 (21.3 g; 0.1 mol) is added dropwise under stirring at 5÷10°C. The mixture is conditioned at 20÷25C for 3–20 h; the reaction is controlled using the 19F NMR technique.
The low–boiling reaction components are then distilled off in vacuum (10 Torr) into a trap (–70°C) and the further distillation of the residue under reduced pressure allows gives a mixture of products of olefin halotrifluoromethyl thiolation that is analyzed using the 19F NMR technique and chromatography–mass spectrometry.
Reaction in the presence of cyclohexene.
The reaction with cyclohexene yields a mixture of products (19F NMR): 50% 10 (δ: –40.2); 20% 11 (δ: –40.4); 30% 12+12’.
(β-Chlorocyclohexyl)trifluoromethyl sulfide (10).
1H NMR δ: 1.3–1.6 (m, 3H, cyclohexyl), 1.6–1.85 (m, 3H, cyclohexyl), 2.15 (m, 1H, cyclohexyl), 2.35 (m, 1H, cyclohexyl), 3.4 (m, 1H, CHSCF3), 4.05 (m, 1H, CHCl); 19F NMR δ: –40.2 (s, SCF3). The mass spectrum (M/Z, reference): 218[M]+, 199[M–F]+, 183[M–Cl]+, 149[M–CF3]+, 141[C3H4SCF3]+, 128[C2H3SCF3]+, 117[C6H10Cl]+, 81[C6H9]+(100%), 79[C6H7]+, 77[C6H5]+, 67[C5H7]+, 59[C2H3S]+, 53[C4H5]+, 45[CHS]+, 39[C3H3]+.
Similarly, a mixture of products is obtained using 2 equivalents of PBr3 in acetonitrile (19F NMR): 84% 11 (δ: –40.4); 16% 12’.
(β-Bromocyclohexyl)trifluoromethyl sulfide (11).
The mass spectrum (M/Z, reference): 262[M]+, 183[M–Br]+, 161[C6H10Br]+, 141[C3H4SCF3]+, 128[C2H3SCF3]+, 81[C6H9]+(100%), 79[C6H7]+, 77[C6H5]+, 67[C5H7]+, 59[C2H3S]+, 53[C4H5]+, 45[CHS]+, 39[C3H3]+.
Reaction in the presence of allyl trifluoroacetamide.
In this case, the reaction does not require catalysis by DMF. A mixture of products is obtained (19F NMR): 10% CF3SSCF3 (δ: –47); 60% MCl 13a (δ: –40.93); 21.6% a-MCl 14a (δ: –42.34); 6.5% MBr 13b (δ: –40.84); 1.9% a-MBr 14b (δ: –42.27).
N-(2-chloro-3-trifluoromethylthio propyl)trifluoroacetamide (13a).
The mass spectrum (M/Z, reference): 290[M+H]+, 270[M–F]+, 254[M–Cl]+, 234[M–Cl–HF]+, 188[M–CF3S]+, 184[M–HCl–CF3]+, 177[M–NHCOCF3]+, 168[C5H5F3NS]+, 152[C5H5F3NO]+(100%), 128[C3H3F3S]+, 126[C3H3F3NO]+, 115[C2H2F3S]+, 78[COCF2]+, 69[CF3]+, 59[COCF]+, 45[CHS]+, 39[C3H3]+.
N-(3-chloro-2-trifluoromethylthio propyl)trifluoroacetamide (14a).
The mass spectrum (M/Z, reference): 290[M+H]+, 270[M–F]+, 254[M–Cl]+, 200[M–HF–CF3]+, 188[M–CF3S]+, 184[M–HCl–CF3]+, 168[C5H5F3NS]+, 152[C5H5F3NO]+(100%), 141[M–HCl–NHCOCF3]+, 126[C3H3F3NO]+, 115[C2H2F3S]+, 78[COCF2]+, 69[CF3]+, 45[CHS]+, 39[C3H3]+.
N-(2-bromo-3-trifluoromethylthio propyl)trifluoroacetamide (13b).
The mass spectrum (M/Z, reference): 334[M+H]+, 314[M–F]+, 254[M–Br]+, 234[M–Br–HF]+, 221[M–NHCOCF3]+, 184[M–HBr–CF3]+, 168[C5H5F3NS]+(100%), 152[C5H5F3NO]+, 141[M–HBr–NHCOCF3]+, 128[C3H3F3S]+, 126[C3H3F3NO]+, 115[C2H2F3S]+, 78[COCF2]+, 69[CF3]+, 59[COCF]+, 45[CHS]+, 39[C3H3]+.
N-(3-bromo-2-trifluoromethylthio propyl)trifluoroacetamide (14b).
The mass spectrum (M/Z, reference): 334[M+H]+, 314[M–F]+, 254[M–Br]+, 234[M–Br–HF]+, 221[M–NHCOCF3]+, 184[M–HBr–CF3]+, 168[C5H5F3NS]+, 152[C5H5F3NO]+(100%), 141[M–HBr–NHCOCF3]+, 128[C3H3F3S]+, 126[C3H3F3NO]+, 115[C2H2F3S]+, 78[COCF2]+, 69[CF3]+, 59[COCF]+, 45[CHS]+, 39[C3H3]+.
Reaction in the presence of allyl acetate.
The reaction with allyl acetate yields a mixture of products (19F NMR): 28% CF3SSCF3 (δ: –47); 42% MCl (δ: –41.05); 11% a-MCl (δ: –42.3); 12% MBr (δ: –40.98); 7% a-MBr (δ: –42.22).
Reaction of CF3SO2Br with two equivalents of PCl3 in the presence of allyl bromide.
1 drop of dimethylformamide is added to a mixture of 27.4 g (0.2 mol) of phosphorus trichloride and 6.05 g (0.05 mol) of allyl bromide and then 21.3 g (0.1 mol) of trifluorometane sulfonyl bromide (1) are added dropwise under stirring at the temperature of 10÷15C. The mixture is conditioned at 20÷25°C for 3 h and distillation using a Vigreux column yields 8 g (80%) of CF3SSCF3 (3).
Reaction of CF3SO2Na with two equivalents of PBr3 in acetonitrile.
34.65 g (0.128 mol) of phosphorus tribromide are added dropwise to a suspension of 10 g (64 mmol) of sodium trifluoromethane sulfinate (15) in 10 ml of acetonitrile under stirring at 5÷10°C. The mixture is stirred at 20÷25°C for 3 h and left overnight, then the products with b.p. below 80°C are distilled off in a cooled receiver (0÷5°C). The distillate obtained is washed with glacial 5% hydrochloric acid and distilled over P2O5 to give 5.2 g (80%) of CF3SSCF3 (3).
References
- Michaelis, Bull.Soc.Chim., 1871, 15, 185.
- H.B. North, J.C. Thomson, JACS, 1918, 40, 774-777.
- E. Krumbiegel, Pat. DE415312C (1925).
- Handbook of Preparative Inorganic Chemistry V.1, 2nd Ed., G. Brauer, Acad. Press Inc., N.Y., London, Trans. from the Germ., 1963, p. 387.
- R. Otto, A. Rossing, Ber., 1891, 24, 3874-3883.
- A.H. Kohlhase, JACS, 1932, 54, 2441-2448.
- W.H. Hunter, B.E. Sorenson, JACS, 1932, 54, 3364-3367.
- W.H. Hunter, B.E. Sorenson, JACS, 1932, 54, 3368-3374.
- A.H. Kohlhase, JACS, 1933, 55, 1291-1292.
- L.I. Zakharkin, G.G. Zhigareva, Zh.Org.Khim., 1973, 9, 891-895.
- A.A. Tyutyunov, L.F. Ibragimova, N.D. Kagramanov, S.R. Sterlin, S.M. Igumnov, XI All-Russian Conference “FLUORINE CHEMISTRY” (devoted to the 110th anniversary of academician I.L. Knunyants), June 26-30, 2016, Moscow Russia, O-43.
- C.T. Ratcliffe, J.M. Shreeve, JACS, 1968, 90, 5403-5408.
- Comprehensive Handbook of Chemical Bond Energies, Yu-Ran Luo, CRC Press, Taylor & Francis Group, Boca Raton, London, New York, 2007.
- T. Mahmood, J.M. Shreeve, Inorg.Chem., 1985, 24, 1395-1398.
- P.R. Resnick, US Pat. №3,560,568 (1971).
- A.A. Tyutyunov, L.F. Ibragimova, N.D. Kagramanov, N.I. Delyagina, V.F. Cherstkov, S.R. Sterlin, S.M. Igumnov, Fluorine notes, 2015, 5(102).
- S.D. Chepik, V.F. Cherstkov, S.R. Sterlin, L.S. German, Bull.Acad.Sci. USSR, div.chem.sci., 1990, 39, 1992-1992.
- S.D. Chepik, V.F. Cherstkov, E.I. Mysov, A.F. Aerov, M.V. Galakhov, S.R. Sterlin, L.S. German, Bull.Acad.Sci. USSR, div.chem.sci., 1991, 40, 2285-2291.
- A. Haas, M. Lieb, Y. Zhang, JFC, 1985, 30, 203-210.
- H.W. Roesky, S. Tutkunkardes, Chem.Ber., 1974, 107, 508-517.
- X. Zhao, A. Wei, B. Yang, T. Li, Q. Li, D. Qiu, K. Lu, JOC, 2017, 82, 9175-9181.
- D.-W. Sun, X. Jiang, M. Jiang, Y. Lin, J.-T. Liu, Eur. JOC, 2017, 24, 3505-3511.
- W.-Y. Huang, L. Lu, Chin.J.Chem., 1992, 10, 268-273.
- A.A. Tyutyunov, L.F. Ibragimova, N.D. Kagramanov, S.R. Sterlin, S.M. Igumnov, Fluorine notes, 2016, 6(109).
Recommended for publication by Prof. S.R. Sterlin
Fluorine Notes, 2018, 117, 1-2
