Received: July 2017
DOI 10.17677/fn20714807.2017.04.03
Fluorine Notes, 2017, 113, 5-6
Asymmetric Construction of Chiral Carbon Centers Featuring the SCF3 Motif: Direct Trifluoromethylthiolation versus Building Block Approach
Hélène Chachignon,1 Evgeniy V. Kondrashov,2 Dominique Cahard*,1
1 UMR 6014 CNRS C.O.B.R.A., Normandie Université, 1 rue Tesnière, 76821 Mont Saint Aignan, France
2 A. E. Favorsky Institute of Chemistry, Siberian Branch of the Russian Academy of Sciences, 1, Favorsky Str., Irkutsk 664033, Russia
Corresponding Author: dominique.cahard@univ-rouen.fr
Abstract: The recent advances in the field of asymmetric synthesis of chiral organofluorine molecules bearing a SCF3 motif attached to a chiral carbon center are presented in this mini-review. Both direct trifluoromethylthiolation and the use of fluorinated building blocks are described.
Keywords: Fluorine – Trifluoromethylthiolation – Asymmetric synthesis – Chirality.
Table of Contents:
1. Introduction
2. Direct trifluoromethylthiolation
2.1 Diastereoselective trifluoromethylthiolation
2.2 Enantioselective trifluoromethylthiolation
2.3 Chiral trifluoromethylthiolation reagents
3. Building block approach
4. Perspectives
1. Introduction
Organofluorine chemistry is a research field of tremendous expansion because the scope of applications of fluorinated molecules is impressively large, impacting all domains of society from pharmaceutical and agrochemical sciences to materials science and energy. In the life sciences area, the role of chirality is crucial for interaction of molecules with the biological target. As a consequence, the stereocontrol at carbon centers featuring a fluorinated motif is an imperative to market enantiomerically pure drugs [1]. From a chemical point of view, the asymmetric synthesis of chiral nonracemic fluorinated compounds is a highly challenging task. Methods for the direct introduction of a fluorine atom and a trifluoromethyl group are manifold and allow high level of stereoselectivity [2]. On the other hand, the asymmetric introduction of emergent fluorochemotypes is still in its infancy. Of the fluorinated motifs exhibiting recent interest, it may be noted that the SCF3 group occupies a place of choice [3]. Indeed, the exceptional lipophilicity that the SCF3 group confers to molecules (Hansch hydrophobic parameter: π= 1.44 versus 0.88 for CF3) and its high electron-withdrawing character (Hammett substituent constants: σm = 0.40, σp = 0.50 versus 0.43, 0.54, respectively for the CF3 group) make it attractive to build novel chemical architectures for the conception of new pharmaceuticals with enhanced capacity to pass cell membranes. Most of the current trifluoromethylthiolated biologically active compounds feature a C(sp2)–SCF3 group mainly on aromatics and two SCF3 molecules are on the market for veterinary use only: Toltrazuril or Baycox® from Bayer and Monepantel or Zolvix® from Novartis. None of the SCF3 compounds is currently marketed for human medication. The development of compounds featuring a C(sp3)–SCF3 group with emphasis for enantiopure ones is of growing interest. In this context, it appears to us timely to review the synthetic approaches for the asymmetric construction of chiral carbon centers featuring the SCF3 motif. This short yet exhaustive review compiled all the papers on this topic till end of June 2017.
2. Direct trifluoromethylthiolation
The most widespread approach for the construction of chiral carbon centers featuring the SCF3 motif is the direct introduction of the SCF3 group on various substrates. Among the strategies employed, most are focused on enantioselective trifluoromethylthiolation by means of chiral catalysts or chiral reagents. Diastereoselective trifluoromethylthiolation is clearly underdeveloped with a single published example. These methods utilize electrophilic trifluoromethylthiolation reagents among those depicted in Figure 1 [4].
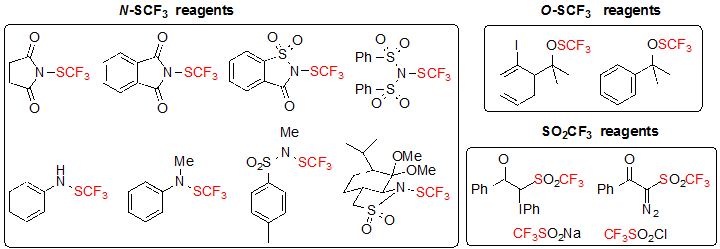
Figure 1. Classes of electrophilic trifluoromethylthiolation reagents
2.1. Diastereoselective trifluoromethylthiolation
The first asymmetric introduction of the SCF3 motif was reported in 2013 by Shen and coworkers. This group proposed the diastereoselective trifluoromethylthiolation of a lithium enolate bearing a chiral Evans phenyloxazolidin-2-one, using a trifluoromethylated thioperoxide as the SCF3 source (Scheme 1) [5].The expected product was obtained with high diastereoselectivity (dr 50:1), albeit in a low yield (20%). However, no further investigation was performed on the reaction conditions. No other example of similar reaction was reported in the literature to this day.
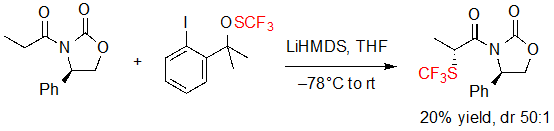
Scheme 1. Diastereoselective trifluoromethylthiolation of a carbonyl compound
2.2. Enantioselective trifluoromethylthiolation
As opposed to diastereoselective trifluoromethylthiolation, reports concerning the enantioselective introduction of the SCF3 moiety on various substrates are more numerous.
Trifluoromethylthiolation of β-ketoesters
In 2013, Shen and coworkers described the first asymmetric organocatalyzed trifluoromethylthiolation of β-ketoesters mediated by quinine-based catalysts. A variety of indanone-derived adamantyl β-ketoesters were readily converted into their (R)- trifluoromethylthiolated derivatives in high yields and excellent enantioselectivities using 20 mol% of quinine as well as 1.2 equivalents of Shen’s trifluoromethylsulfenate reagent in toluene at 40 °C (Scheme 2) [6].
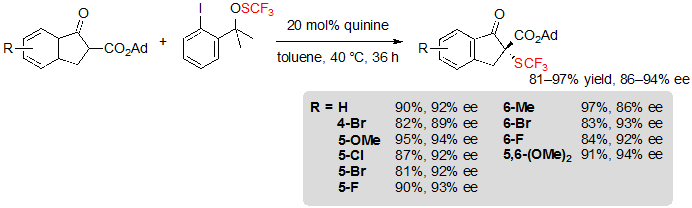
Scheme 2. Asymmetric trifluoromethylthiolation of indanone-based adamantyl β-ketoesters
The enantioselectivity proved to be particularly sensitive to the steric hindrance of the ester group; indeed, ee values dropped dramatically when performing the reaction on methyl or ethyl esters. On the contrary, the nature and position of the benzene ring substituents appeared to have little impact on the proceeding of the reaction as similar results were obtained with substrates bearing electron-withdrawing or electron-donating groups. The conditions showed as well good compatibility with a cyclopentanone-derived β-ketoester, which furnished the corresponding product in 95% yield with 94% ee. On the other hand, in the case of tetralone-derived substrates, very low conversions (<5%) were observed. Mechanistic studies were carried out in order to rationalize this phenomenon. Preliminary formation of a N-SCF3 quaternary ammonium produced from quinine and the SCF3 source was first considered by the authors. However, no newly formed species could be observed when monitoring the reaction of these two reagents by 19F NMR in the standard conditions, so this hypothesis was ruled out (Scheme 3).
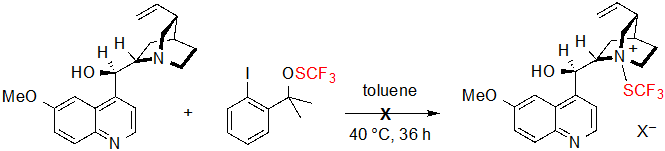
Scheme 3. Expected reaction between quinine and the trifluoromethylthiolation reagent
Consequently, a dual activation of both the substrate and the trifluoromethylthiolation agent by means of hydrogen bonding with the quinine was proposed (Scheme 4). Such hypothesis is consistent with the fact that the use of other cinchona alkaloids, which do not feature a hydroxyl group, resulted in extremely low conversions. Moreover, considering that tetralone-derived β-ketoesters form non-planar enolates, which probably cannot fit in this restrictive model nor, therefore, generate the dual-activated intermediate, this mechanism can justify their lack of reactivity.
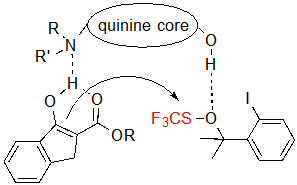
Scheme 4. Dual activation of the substrate and the trifluoromethylthiolation reagent
As a result, some modifications of the reaction conditions were necessary to address this issue. Among others, replacing quinine by a quininium based phase-transfer catalyst greatly contributed to reach higher yields. Hence, five SCF3 tetralone-based β-ketoesters could be obtained in excellent yields and moderate enantioselectivities (Scheme 5). 1-Benzosuberone derivatives as well as cyclohexanone-based substrates were also compatible with the modified reaction conditions. Interestingly enough, said conditions did not allow a satisfying conversion of five-membered ring β-ketoesters.
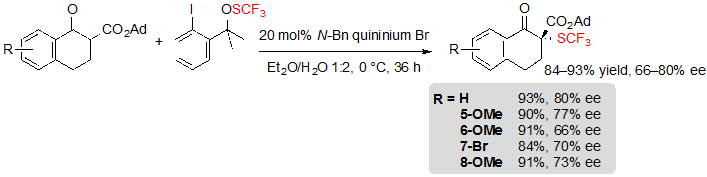
Scheme 5. Asymmetric trifluoromethylthiolation of tetralone-based adamantyl β-ketoesters
Acyclic substrates proved to be the main limitation of these reactions, as they showed poor reactivity when using either of the two proposed quinine-based catalysts.
The asymmetric trifluoromethylthiolation of β-ketoesters did not only attract the interest of Shen’s group. Indeed, Rueping and coworkers simultaneously reported a similar methodology, also based on the use of cinchona alkaloid catalysis [7].This time, N-(trifluoromethylthio)phthalimide was employed as the electrophilic SCF3 source. Using 10 mol% of quinidine in dichloromethane at –75 °C, various (S)-indanone-derived trifluoromethylthiolated β-ketoesters were produced in excellent yields and enantioselectivities (Scheme 6).
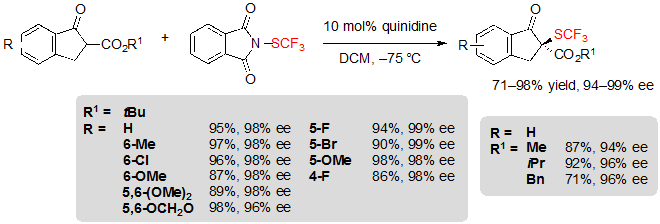
Scheme 6. Asymmetric trifluoromethylthiolation of indanone-based β-ketoesters
It is worth underlining that, in this case, the size of the ester group had a very limited influence on both the yield and the enantioselectivity of the reaction. Furthermore, a tetralone-based substrate could also be converted into its SCF3 analogue, albeit in moderate yield (46% yield, 95% ee), by increasing the temperature to 0 °C and lengthening the reaction time to seven days.
Cyclopentenone-derived tert-butyl β-ketoesters were also compatible with the reaction conditions, offering the corresponding products in good yields and high enantioselectivities (Scheme 7).
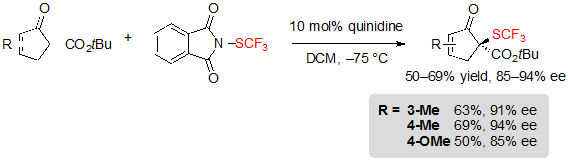
Scheme 7. Asymmetric trifluoromethylthiolation of cyclopentenone-based β-ketoesters
Finally, it was also possible to efficiently access the (R)-enantiomer by switching quinidine for its pseudo enantiomer quinine (Scheme 8).
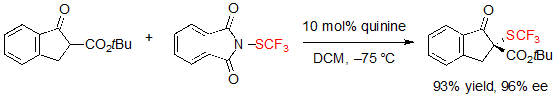
Scheme 8. Access to (R)-SCF3 indanone-based β-ketoester
In addition to these organocatalyzed approaches, Gade and coworkers described a method based on metal-catalysis for conducting the same kind of reaction. The association of a copper-boxmi complex and Shen’s trifluoromethylsulfenate reagent allowed to obtain numerous trifluoromethylthiolated cyclic β-ketoesters in excellent yields and with high enantioselectivities (Scheme 9) [8]. The reaction was compatible with indanone-, tetralone-, cylcopentenone- as well as cyclohexenone-derived β-ketoesters, and diverse functional groups such as methoxy, methyl or halogens were well-tolerated. Once again, the bulkiness of the ester group had little impact on the enantioselectivity, except when performing the reaction on tetralone derivatives.
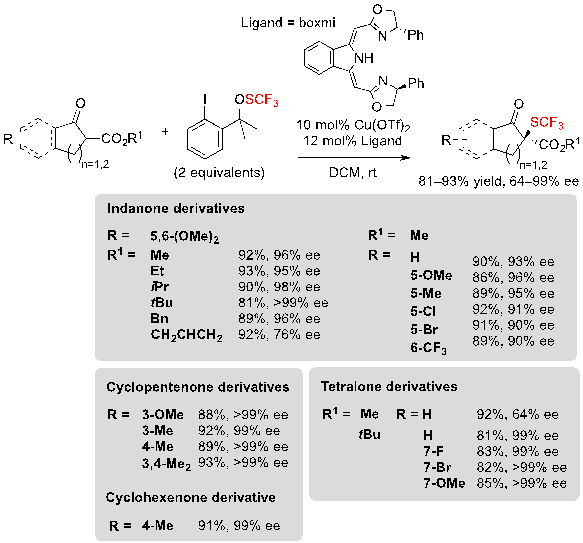
Scheme 9. Copper-catalyzed trifluoromethylthiolation of various β-ketoesters
As for the mechanism of the reaction, organocatalysis by the sole action of the ligand was quickly excluded, as no product was formed in absence of copper salts. Moreover, a potential direct reaction between the SCF3 reagent and the copper complex was not witnessed either by EPR spectroscopy nor by 19F NMR spectroscopy. Considering these information experiments, the authors proposed the pathway described in Scheme 10. In the first place, both carbonyl oxygen atoms of the β-ketoester coordinate to the copper-boxmi complex. The obtained intermediate A is then deprotonated by triflate anion to afford the ester enolate B, on which the SCF3 group can be introduced from the trifluoromethylthiolation reagent via a triflic acid activation. The final product is produced from intermediate C after an ultimate step of de-coordination. The enantioselectivity of the process supposedly emerges from the fact that the Si face of enolate B is shielded by the phenyl ring of the boxmi ligand, and that, consequently, electrophilic SCF3 preferentially approaches from the Re face.
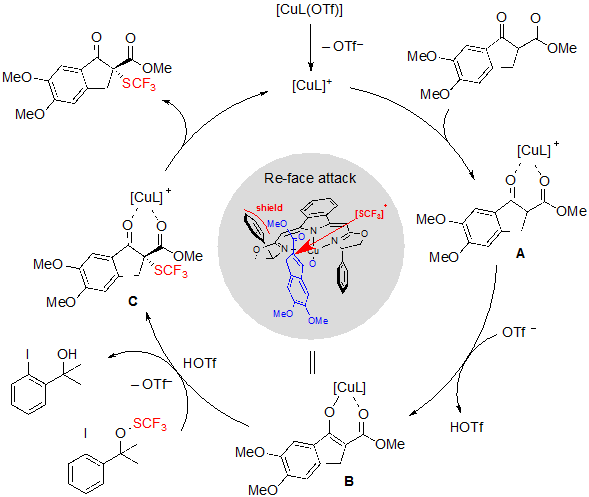
Scheme 10. Mechanism of the Cu-catalyzed trifluoromethylthiolation of cyclic β-ketoesters
Du and coworkers were also interested in the asymmetric introduction of the SCF3 motif on 1,3-dicarbonyls. In 2017, they proposed the enantioselective trifluoromethylthiolation of a cycloalkenone thanks to N-trifluoromethylthiosuccinimide and a squaramide catalyst derived from hydroquinine and D-glucopyranose (Scheme 11) [9].
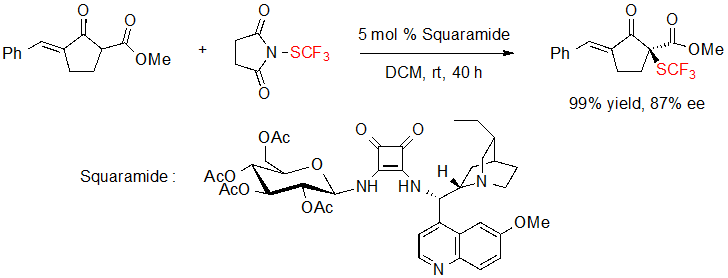
Scheme 11. Enantioselective trifluoromethylthiolation of a cycloalkenone by a squaramide catalyst
Such substrates could also be reacted with 2-mercaptobenzaldehyde derivatives in a one-pot procedure to furnish SCF3 spiro cyclopentanone-thiochromanes through a sulfur-Michael/aldol cascade reaction (Scheme 12).
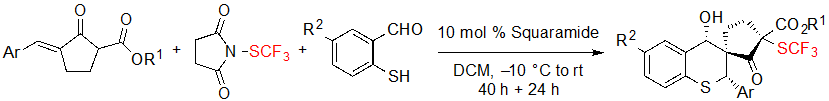
Scheme 12. One-pot synthesis of SCF3 spiro cyclopentanone-thiochromanes
Interestingly, a cyclohexenone derivative was efficiently converted into its SCF3 analogue using slightly tuned conditions but could not take part in the cascade reaction (Scheme 13).
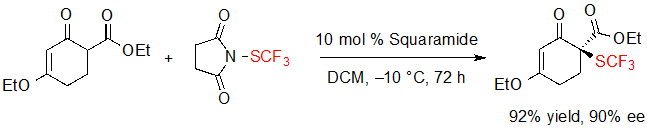
Scheme 13. Enantioselective trifluoromethylthiolation of a cyclohexenone derivative
Trifluoromethylthiolation of oxindoles
Oxindoles were also privileged substrates for the enantioselective introduction of the SCF3 moiety. Rueping and coworkers first reported a procedure allowing the asymmetric trifluoromethylthiolation of oxindoles using similar conditions as those they proposed for β-ketoesters (Scheme 14) [10].Nonetheless, quinidine had to be replaced by a bis-cinchona alkaloid catalyst, (DHQD)2Pyr, to obtain high enantioselectivities. Various 3-aryl Boc-protected oxindoles, substituted by different motifs on the aryl as well as on the fused-phenyl ring of the heterocycle, furnished indifferently the associated products in good to excellent yields and enantioselectivities.
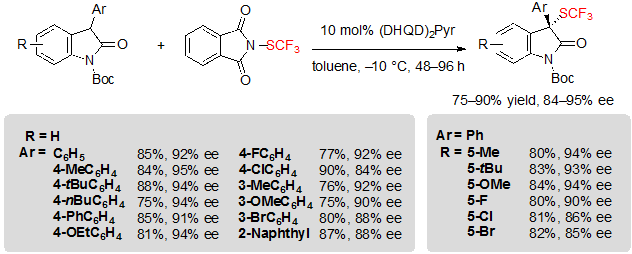
Scheme 14. Asymmetric trifluoromethylthiolation of 3-aryl oxindoles
Shortly after, Tan, Liu and coworkers described a closely related methodology, although based on the in situ generation of an electrophilic trifluoromethylthiolation reagent from readily available AgSCF3 and trichloroisocyanuric acid (TCCA) (Scheme 15) [11].The reaction conditions were compatible with 3-aryl oxindoles with various substitution patterns, and offered the expected products in moderate to excellent yields and with high enantioselectivities after a step of one-pot deprotection.
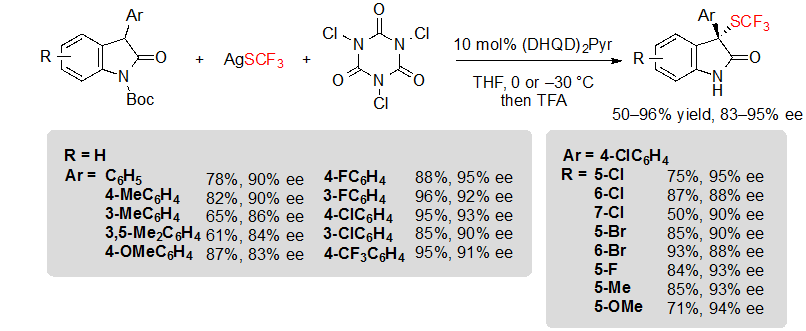
Scheme 15. Asymmetric trifluoromethylthiolation of 3-aryl oxindoles with in situ generation of the electrophilic SCF3 donor
Experiments carried out for the identification of the reactive electrophilic SCF3 showed that bis(trifluoromethyl)disulfide was generated in situ, and took part in the conversion of the substrate. However, formation of the trifluoromethylthiolated product also occurred with similar enantioselectivity without said disulfide, albeit in presence of insoluble species issued from the reaction of TCCA and AgSCF3. Unfortunately, these species could not be identified (Scheme 16).
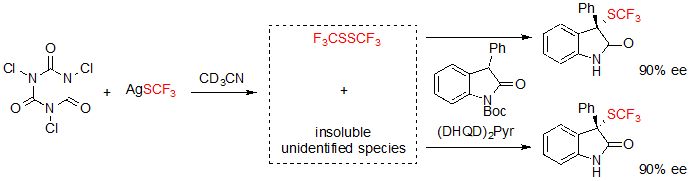
Scheme 16. Investigation on the active species of the reaction
Shen and coworkers finally proposed an alternative methodology, although based on a similar concept, with the noteworthy advantage of giving access to unprecedented trifluoromethylthiolated 3-alkyl oxindoles in excellent yields and with high enantioselectivities (Scheme 17) [12]. To achieve such results, it was necessary to use 1.2 equivalents of Shen’s trifluoromethylsulfenate reagent along with 20 mol% of quinine, in diethyl ether, at 35 °C. The nature of the trifluoromethylthiolation reagent proved to be truly influential on the reaction; indeed, replacing it by N-(trifluoromethylthio)phthalimide triggered an important decrease of the enantioselectivity. Unfortunately, the reaction proceeded less efficiently with 3-aryloxindoles, offering the associated products with slightly lower enantioselectivities. On the bright side, it was found that other protecting groups than Boc, such as CO2Ad, could be tolerated without impacting drastically the ee values.
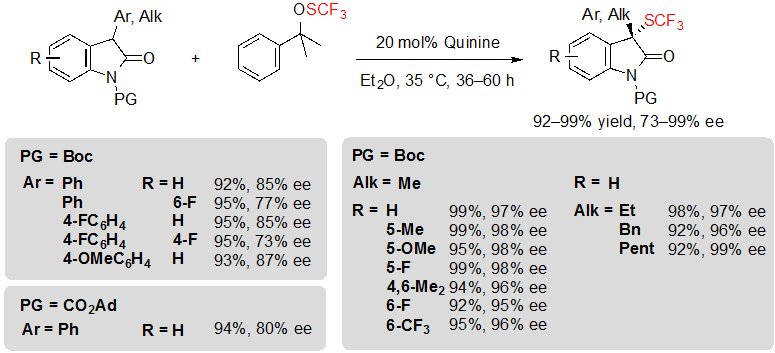
Scheme 17. Asymmetric trifluoromethylthiolation of 3-aryl and 3-alkyl oxindoles
Trifluoromethylthiolation reactions toward dithioketals
The group of Zhou has been interested in the synthesis of dithioketal motifs, as they display promising potential in biopharmaceutical chemistry. In this context, they described the asymmetric introduction of the SCF3 group onto 1-indanone-derived thioethers using 1.2 equivalents of N-(trifluoromethylthio)succinimide and 10 mol% dihydroquinine in dichloromethane at –20 °C. After six days, the corresponding SCF3 products were isolated with excellent enantioselectivities, albeit in moderate yields (Scheme 18) [13].
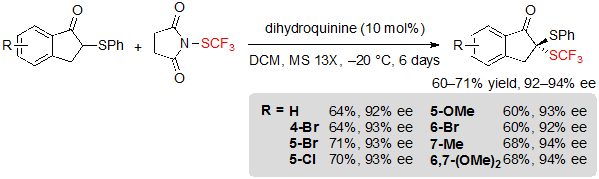
Scheme 18. Asymmetric trifluoromethylthiolation of 1-indanone-derived thioethers
Interestingly, it was also possible to access more diverse dithioketals issued from oxindole, 2-indanone or 3-benzofuranone with similarly moderate yields and good ee values (Scheme 19).
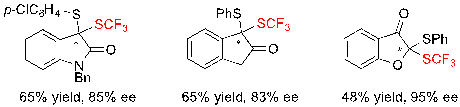
Scheme 19. Access to structurally diverse dithioketals
Trifluoromethylthiolation of aldehydes
During their research focusing on the trifluoromethylthiolation of aldehydes, Wu, Sun and coworkers attempted to extend their methodology by developing an asymmetric version of it (Scheme 20) [14]. Disappointingly, only 11% ee value was measured when performing the reaction in the presence of the Hayashi-Jørgensen catalyst. To this day, no further investigation on this reaction has been reported.
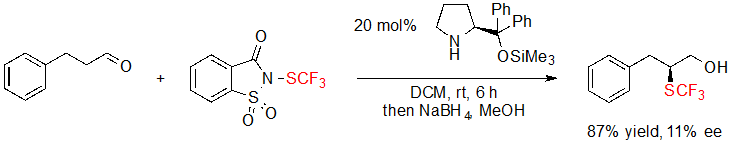
Scheme 20. Asymmetric α-trifluoromethylthiolation of an aldehyde
Cascade trifluoromethylthiolation / cyclization from functionalized alkenes
Recently, Zhao and coworkers studied the challenging enantioselective trifluoromethylthiolation of alkenes followed by a cyclization step for the synthesis of chiral lactones. For this purpose, a bifunctional chiral sulfide or selenide catalyst was used with a newly developed trifluoromethylthiolation reagent, (PhSO2)2N–SCF3, analogue of the well-known fluorinating agent NFSI. Thus, using 1.5 equivalents of said reagent, 0.5 equivalents of triflic acid and 20 mol% of a chiral indane-based sulfide in dichloromethane at 0 °C, a variety of 4-aryl substituted (E)-but-3-enoic acids could be converted into the corresponding lactones through a cascade trifluoromethythiolation / lactonization reaction (Scheme 21) [15,16]. A slight decrease of enantioselectivity was observed for substrates with aryls carrying an electron-donating group in para position. Similarly, hindered alkenes, with 2-substituted aryl moiety, led to lesser ee values. On the other hand, when electron-withdrawing groups were introduced on para or meta positions of the aryl, the yield was the most impacted and decreased down to 55%. The aryl substituent could be switched to a benzyl group without much consequence; however, replacing it by long alkyl chains led to mixture of diastereoisomers and moderate enantioselectivities.
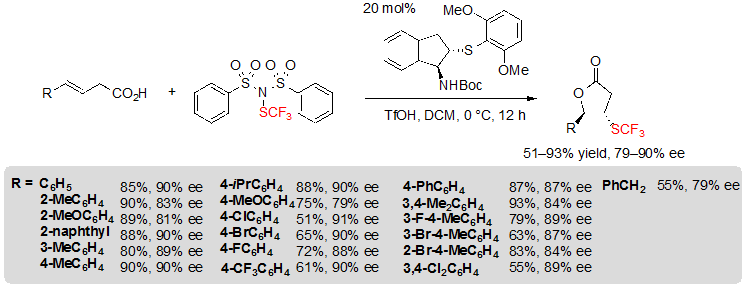
Scheme 21. Asymmetric trifluoromethylthiolating lactonization of alkenes
As for the mechanism of the reaction, the authors proposed the pathway represented on scheme 22. The Boc-protected catalyst first reacts with triflic acid to form the corresponding triflate A. The SCF3 cation is then bound to the catalyst to form the intermediate sulfenylium ion B via a triflic acid activation. In the presence of an olefinic carboxylic acid, intermediate B is readily converted into the trifluoromethylthiiranium ion C, which is finally attacked by the oxygen of the carboxylic group to yield the expected product and regenerating the catalyst in the process. This mechanism enlightens the utility of the bifunctional catalyst, which is not only able to bind to the SCF3 cation through the sulfur atom but also to dispose the carboxylic group for the cyclisation step. Interestingly, structurally close catalysts featuring a selenium instead of a sulfur atom could also be used, but lower enantioselectivities were achieved. This can be explained by the smaller size of the sulfur atom, which led to enhanced steric hindrance around the SCF3 cation, and consequently resulted in better stereocontrol.
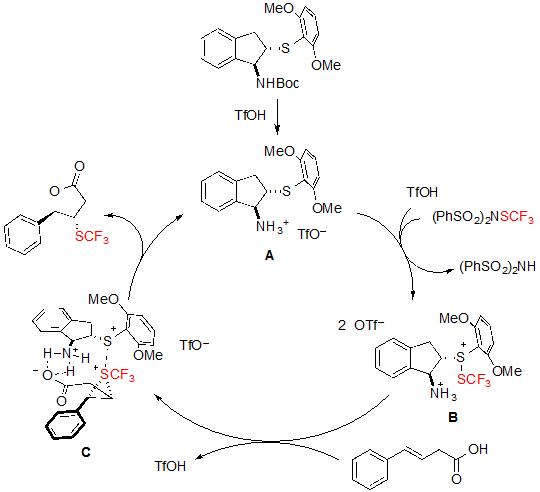
Scheme 22. Mechanism of the asymmetric trifluoromethylthiolating lactonization of alkenes
In the context of their work on trifluoromethylthiolating cyclization of alkenes, Zhao and coworkers also described an access to saturated SCF3 azaheterocycles based on a similar concept (Scheme 23) [17]. To serve this purpose, the same trifluoromethylthiolation reagent, (PhSO2)2N–SCF3, was selected and an indane-based chiral selenide catalyst bearing a triflyl group was used. As it was already the case in Zhao’s previous work, steric hindrance and electronic effects of the catalyst were the key parameters for fine tuning in order to obtain high ee values.
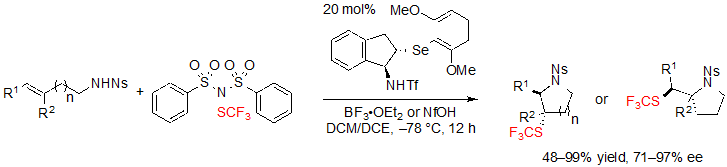
Scheme 23. Asymmetric trifluoromethylthiolating cyclization of (E)-alkenes toward saturated SCF3 azaheterocycles
Thus, using 1.5 equivalents of (PhSO2)2NSCF3, 20 mol% of the selenide catalyst and 1.0 equivalent of BF3·OEt2 in a 1:1 mixture of dichloromethane and dichloroethane at –78 °C, various (E)-alk-3-ene-1-sulfonamides furnished the associated products in high yields, dr and ee values (Scheme 24). Alkyl- and aryl-substituted alkenes were equally well-tolerated. Arylalkenes with electron-withdrawing groups like halogens were, however, less easily converted and necessitated a rise of the reaction temperature to afford high yields. On the other hand, (Z)-isomers did not react.
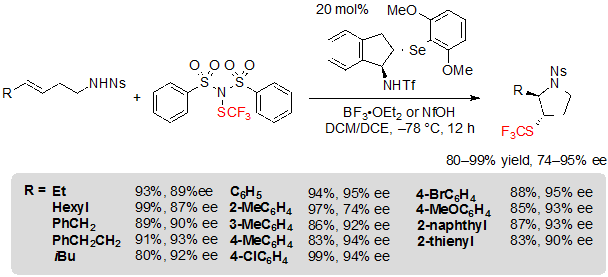
Scheme 24. Asymmetric trifluoromethylthiolating cyclization of (E)-alkenes toward saturated five-membered SCF3 azaheterocycles
Alk-4-ene-1-sulfonamides proved to be more challenging substrates; indeed, competition between endo and exo cyclization was observed and the nature of the obtained product was more dependent on the substitution of the starting material. The exo product was predominantly obtained in the case of terminal, alkyl, and trisubstituted alkenes in moderate to excellent yields and enantioselectivities (Scheme 25).
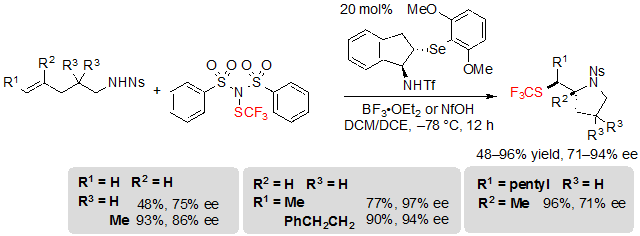
Scheme 25. Asymmetric trifluoromethylthiolating exo cyclization of (E)-alk-4-enes toward pyrrolidines
The formation of the exo product occurred as the main pathway when performing the reaction with a phenyl substituent on the alkene (Scheme 26).
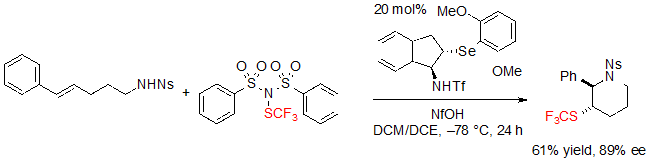
Scheme 26. Asymmetric trifluoromethylthiolating endo cyclization of a (E)-alk-4-ene toward a saturated six-membered SCF3 azaheterocycle
2.3. Chiral trifluoromethylthiolation reagents
In 2017, Shen and coworkers proposed the use of optically pure trifluoromethylthiolation reagents for the enantioselective trifluoromethylthiolation [18]. (1S)-(–)-N-Trifluoromethylthio-2,10-camphorsultam derivatives proved to be the best candidates to perform this type of reaction. N-Trifluoromethylthio-oxazolidinone derivatives were also evaluated, but gave lower enantioselectivities (Scheme 27).
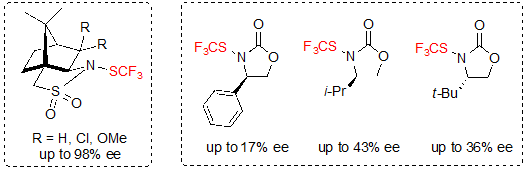
Scheme 27. Chiral trifluoromethylthiolation reagents (ee values for the reaction with β-ketoesters)
These reagents were compatible with various substrates. For example, in the presence of 0.1 equivalents of K2CO3 in tetrahydrofuran at –40 °C, five and six-membered ring β-ketoesters afforded the corresponding SCF3 products in good to excellent yields with high enantioselectivities, up to 98% (Scheme 28). Several functional groups (R) and diverse ester groups (R1) were well tolerated. However, it was necessary to increase the temperature to reach a satisfying conversion when performing the reaction on substrates carrying electron-withdrawing R groups.
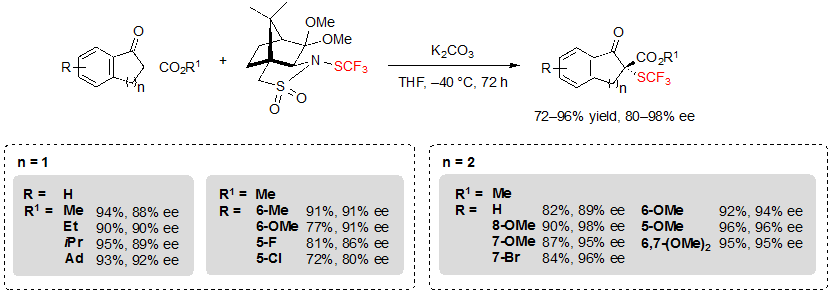
Scheme 28. Asymmetric trifluoromethylthiolation of indanone- and tetralone-based cyclic β-ketoesters
Similarly, a variety of 3-aryloxindoles were readily converted into their trifluoromethylthiolated analogues after a slight modification of the reaction conditions. Notably, K2CO3 was replaced by Cs2CO3 and tetrahydrofuran by diethyl ether (Scheme 29). Generally, the reaction conditions were once again efficient regardless of the functional groups carried by the aryl group or the nature of the protecting group linked to the nitrogen. However, in the unique case of the oxindole carrying a cyanomethyl substituent at the para position of its 3-aryl ring, a drop of enantioselectivity was observed. This issue could nonetheless be solved by replacing the trifluoromethylthiolating reagent to another camphorsultam analogue. Said reagent also allowed to reach higher ee values for already evaluated substrates, and even permitted to perform the reaction on a 3-methyl oxindole with moderate enantioselectivity.
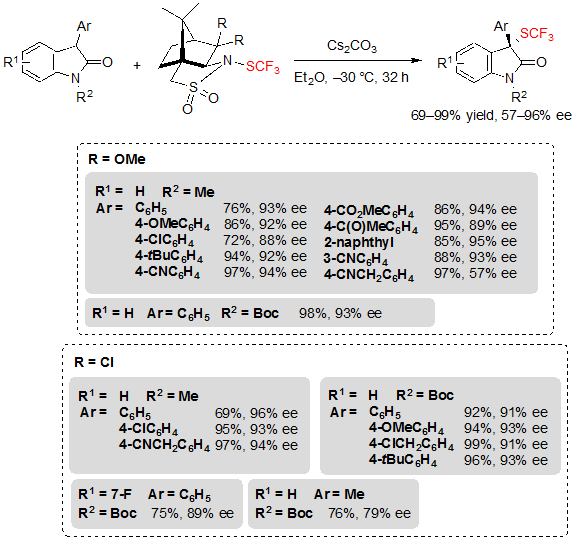
Scheme 29. Asymmetric trifluoromethylthiolation of 3-aryloxindoles
Finally, it was also possible to access structurally new chiral 2-SCF3-2-aryl benzofuran-2(3H)-ones in moderate to high yields and with ee values in the range 64–92% ee (Scheme 30).
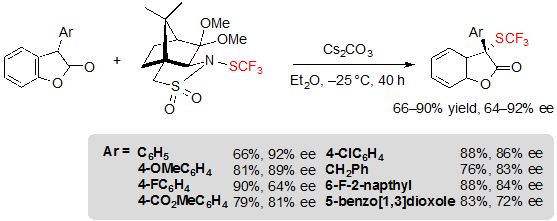
Scheme 30. Asymmetric trifluoromethylthiolation of 2-aryl benzofuran-2(3H)-ones
3. Building block approach
In comparison with direct enantioselective trifluoromethylthiolation methods, the building block approach for construction of chiral centers bearing the SCF3 group is little widespread. At the present time, only two recent reports describing stereocontrolled formation of chiral SCF3 compounds can be found.
The first approach, implemented by Wang and coworkers is a variation of the Doyle-Kirmse reaction [19]. It proceeds through an enantioselective [2,3]-sigmatropic rearrangement of a sulfonium ylide that is generated from allyl or propargyl trifluoromethyl sulfides and a metal carbene species issued from the reaction of a diazoester and a metal catalyst bearing chiral ligands. This methodology was first developed using a rhodium (II) catalyst, Rh2(S-DOSP)4, in n-pentane, at –30 °C. A wide range of aromatic diazoesters were submitted to these reaction conditions, giving access to the corresponding chiral (R)-trifluoromethyl sulfides in good to excellent yields with high ee values (Scheme 31). Interestingly, the bulkiness of the ester group displayed an important influence on the enantioselectivity of the reaction; indeed, ee values dropped as the steric bulk of the ester increased. Electronic effects also proved to be of importance; diazoesters bearing aryls with electron-donating groups were converted with diminished yields and enantioselectivities. This phenomenon may be related to the lesser stability of such compounds, which therefore are more prone to be consumed by side-reactions like dimerization.
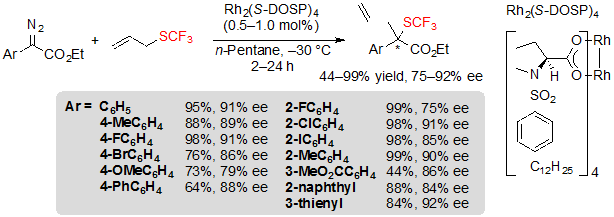
Scheme 31. Rh-catalyzed [2,3] sigmatropic rearrangement from aryl diazoesters and allyl trifluoromethylsulfide
The reaction could also be performed efficiently on vinyl-diazoacetates and higher ee values were actually achieved with these substrates (Scheme 32).
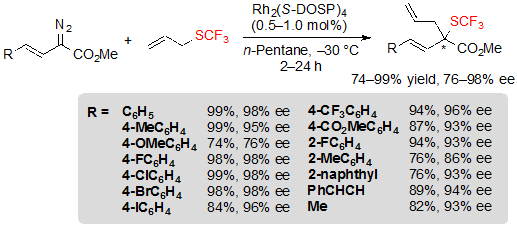
Scheme 32. Rh-catalyzed [2,3] sigmatropic rearrangement from vinyl diazoesters and allyl trifluoromethylsulfide
Finally, it was also possible to replace the allyl trifluoromethyl sulfide by propargyl trifluoromethyl sulfide and to react it with similar aryl- and vinyl-diazoesters in order to access the corresponding allenyl sulfides. The reaction proceeded in moderate to good yields as well as enantioselectivities (Scheme 33).
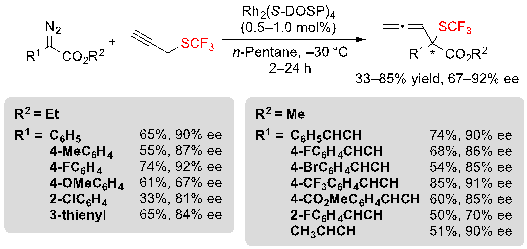
Scheme 33. Rh-catalyzed [2,3] sigmatropic rearrangement from diazoesters and propargyl trifluoromethylsulfide
Copper(I) catalysts, which present the advantage to be more cost-effective than rhodium complexes, were also evaluated. When using Cu(CH3CN)4PF6 associated with a chiral ligand, the reaction showed good efficiency and afforded the expected products with essentially identical enantioselectivity (Scheme 34). The reaction suffered from similar limitations as with rhodium catalysis: diazoesters bearing electron-rich aromatic rings gave low yields and moderately high enantioselectivities. On the other hand, in these conditions, vinyl-diazoacetates furnished low ee values, and conversion of propargyl sulfides was especially slow.
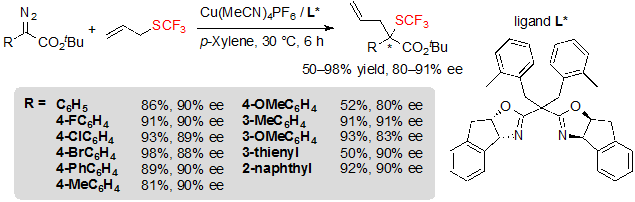
Scheme 34. Cu-catalyzed [2,3] sigmatropic rearrangement from diazoesters and allyl trifluoromethylsulfide
As mentioned earlier in the text, the considered reaction is an asymmetric example of Doyle-Kirmse reaction. The first step of said reaction consists in the formation of a chiral metal carbene, followed by its coordination with the sulfur atom of the allyl sufide. The obtained sulfonium ylide then underwent a concerted [2,3]-sigmatropic rearrangement, transferring the chirality from sulfur to carbon (Scheme 35). The key step in respect to enantioselectivity is the formation of the free ylide. The hypothesis that the reaction might process through a Rh(II)-associated ylide was ruled out by several control experiments.
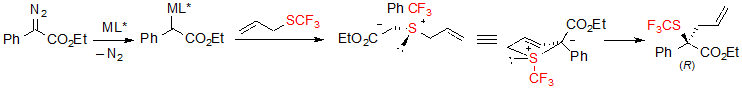
Scheme 35. Reaction mechanism
Zhao and coworkers focused on a different type of building block and described asymmetric organophosphine-catalyzed Mannich-type reactions on N-Boc imines giving access to tetrasubstituted carbon stereocenters featuring a cyano and a SCF3 group [20]. In this study, aryl 2-(trifluoromethylthio)acetonitriles were selected as substrates. The best results were obtained when using 1.0 equivalent of SCF3 building block, 2.0 equivalents of Boc-protected imines, 10 mol% of chiral phosphine and 10 mol% of methyl acrylate. The expected products were obtained with moderate dr (up to 78:22) and moderate to high ee values, consequently the yields related to both diastereomers were limited, up to 68% for the major diastereomer (Scheme 36). Similar yields were obtained indifferently to the substitution pattern of the substrates. As for enantioselectivity, electron-donating groups carried by the Ar group of the imine contributed to increase perceptibly the obtained ee values, while electron-withdrawing groups such as halogens gave more disappointing results. The imine protecting group also proved to be quite influential. Indeed, the reaction did not proceed when using p-methoxyphenyl, p-toluenesulfonyl, 1,1-diphenylmethenyl or diphenylphosphinyl, while it worked well with N-CBz imine, although with lower ee value.
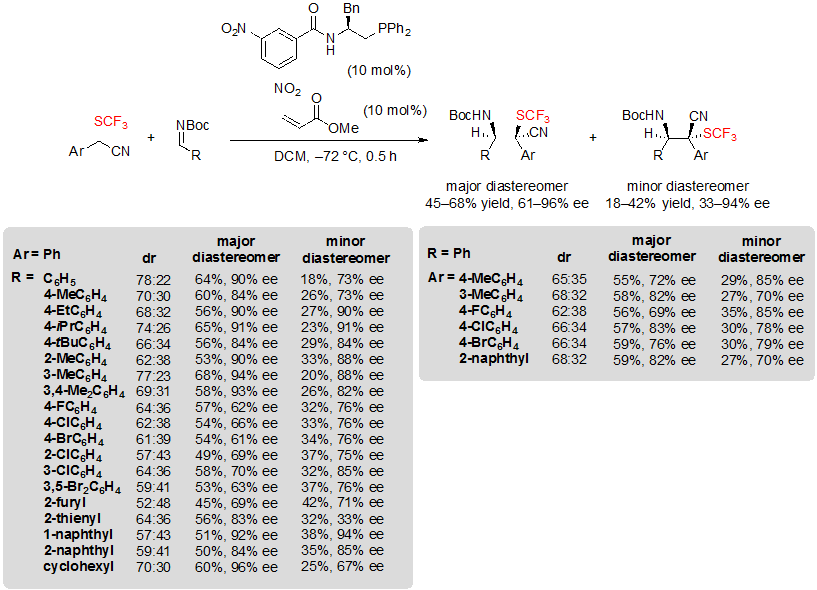
Scheme 36. Formation of CF3S-containing tetrasubstituted carbon stereocenters through enantioselective Mannich-type reactions
Concerning the mechanism of the reaction, the authors proposed the pathway represented on scheme 37. First, the two components of the catalytic system – the chiral phosphine and methyl acrylate – form the zwitterionic adduct A, as confirmed by ESI-MS, that acts as mild chiral BrØnsted base. This phosphonium enolate A deprotonates the SCF3-substrate to generate the ion-pair B, which is then able to attack the Boc-imine, furnishing intermediate D via transition-state C. A proton exchange then occurs between the SCF3 building block and intermediate D producing the mixture of diastereomers and regenerating the species B. This mechanism enlightens the importance of the nitrile group on the SCF3 substrate. Indeed, the nitrile is necessary for the efficient processing of the reaction because it favours greatly the deprotonation of the substrate. It is noteworthy that the reaction could not proceed when replacing this cyano group by an ester moiety.
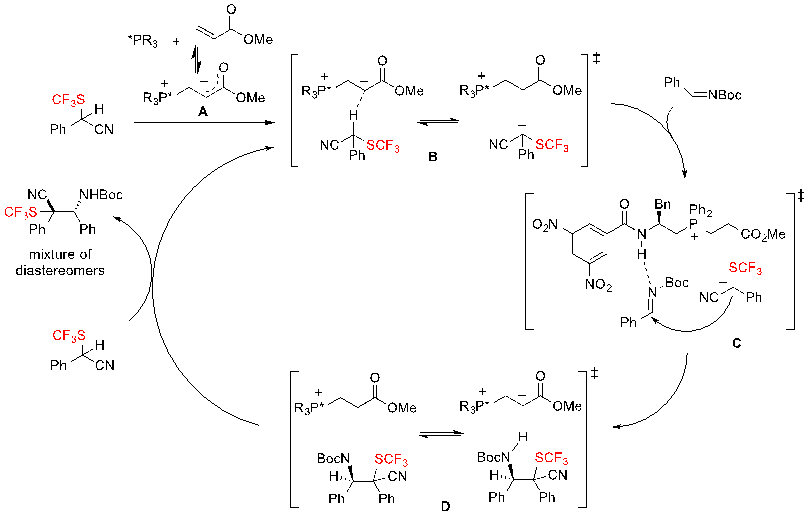
Scheme 37. Proposed mechanism for the enantioselective Mannich-type reaction
4. Perspectives
The asymmetric synthesis of trifluoromethylthiolated molecules is a fledgling area of research in organofluorine chemistry. Since 2013, approaches for the direct introduction of the electrophilic SCF3 motif through organocatalysis, metal-catalyzed reactions, or by means of chiral reagents were described in high-impact publications. More recently, the use of building blocks featuring the SCF3 motif were reported for the stereocontrolled formation of chiral SCF3 compounds. At this stage, research efforts still need to be made in two directions: (i) the discovery of alternative more efficient, cheap, and easy to handle reagents acting as SCF3 donors; (ii) the development of new asymmetric syntheses by innovative approaches. These new tools will allow to reach new SCF3 molecular architectures and ever higher stereoselectivities. We are confident that the current state-of-the-art detailed in this review should stimulate further developments in the field of chiral trifluoromethylthiolated molecules in medicinal applications, material sciences, and other domains of our daily life.
Acknowledgement.
Our research was supported by the Centre National de la Recherche Scientifique (CNRS), University of Rouen, INSA Rouen, and Labex SynOrg (ANR-11-LABX-0029). H. C. thanks the French “Ministère de la Recherche et de l’Enseignement Supérieur” for a doctoral fellowship. E. K. is grateful to the French Embassy in Moscow for a Metchnikov research grant.
References
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa, H. Liu, Chem. Rev. 2016, 116, 422-518.
- a) V. Bizet, T. Besset, J.-A. Ma, D. Cahard, Curr. Top. Med. Chem. 2014, 14, 901-940; b) J.-A. Ma, D. Cahard, Chem. Rev. 2008, 108, PR1-PR43; c) J. Nie, H.-C. Guo, D. Cahard, J.-A. Ma, Chem. Rev. 2011, 111, 455-529.
- a) F. Toulgoat, T. Billard, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds (Eds.: F. R. Leroux, A. Tressaud), Elsevier, 2017, pp. 141-179; b) X. H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 2015, 115, 731-764.
- H. Chachignon, D. Cahard, Chinese J. Chem. 2016, 34, 445-454.
- X. Shao, X. Wang, T. Yang, L. Lu, Q. Shen, Angew. Chem., Int. Ed. 2013, 52, 3457-3460.
- X. Wang, T. Yang, X. Cheng, Q. Shen, Angew. Chem., Int. Ed. 2013, 52, 12860-12864.
- T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei, M. Rueping, Angew. Chem., Int. Ed. 2013, 52, 12856-12859.
- Q.-H. Deng, C. Rettenmeier, H. Wadepohl, L. H. Gade, Chem. Eur. J. 2014, 20, 93-97.
- B.-L. Zhao, D.-M. Du, Org. Lett. 2017, 19, 1036-1039.
- M. Rueping, X. Liu, T. Bootwicha, R. Pluta, C. Merkens, Chem. Commun. 2014, 50, 2508-2511.
- X. L. Zhu, J. H. Xu, D. J. Cheng, L. J. Zhao, X. Y. Liu, B. Tan, Org. Lett. 2014, 16, 2192-2195.
- T. Yang, Q. Shen, L. Lu, Chinese J. Chem. 2014, 32, 678-680.
- K. Liao, F. Zhou, J. S. Yu, W. M. Gao, J. Zhou, Chem. Commun. 2015, 51, 16255-16258.
- L. Hu, M. Wu, H. Wan, J. Wang, G. Wang, H. Guo, S. Sun, New J. Chem. 2016, 40, 6550-6553.
- X. Liu, R. An, X. Zhang, J. Luo, X. Zhao, Angew. Chem., Int. Ed. 2016, 55, 5846-5850.
- J. Luo, X. Liu, X. Zhao, Synlett 2017, 28, 397-401.
- J. Luo, Y. Liu, X. Zhao, Org. Lett. 2017, 19, 3434-3437.
- H. Zhang, X. Leng, X. Wan, Q. Shen, Org. Chem. Front. 2017, 4, 1051-1057.
- Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W.-D. Chu, Y. Zhang, J. Wang, Nat Chem 2017, DOI: 10.1038/nchem.2789.
- G. Zhao, L. Xu, C. Zheng, H. Wang, Adv. Synth. Catal. 2017, DOI: 10.1002/adsc.201700321.
Recommended for publication by Prof. S. M. Igumnov
Fluorine Notes, 2017, 113, 5-6
