Received: July 2017
DOI 10.17677/fn20714807.2017.04.02
Fluorine Notes, 2017, 113, 3-4
Fluorination of some functionalized cycloalkenes through epoxidation and oxirane opening with Deoxofluor or XtalFluor-E
Attila Márió Remete,1 Ferenc Fülöp,1,2 Loránd Kiss1*
1Institute of Pharmaceutical Chemistry, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
2MTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, H-6720 Szeged, Eötvös u. 6, Hungary
Email: kiss.lorand00@gmail.com
Graphical abstract:

Abstract: A convenient, simple synthetic method is described for the synthesis of some fluorine-containing, highly functionalized cycloalkane derivatives. The synthetic protocol involves the stereoselective epoxidation of selected substituted cyclohexenes as model compounds, followed by regioselective opening of epoxides under various conditions with fluorinating reagents Deoxofluor and XtalFluor-E.
Keywords:cycloalkanes,oxirane, fluorination, selectivity
Introduction
The epoxidation of a carbon-carbon double bond and the ring opening of resulting oxiranes with a wide range of nucleophiles is a common method to introduce various functional groups onto the skeleton of a certain organic scaffold [1-5].
Because of the high importance of fluorinated biomolecules in drug design, the introduction of one or more fluorine atoms into an organic molecule has attracted high interest over the past ten years [6-10]. In view of the significance of a fluorine atom in an organic molecule, epoxide opening with fluoride, without doubt, is a highly useful approach for the introduction of a fluorine atom into a certain molecule [11-13]. Numerous synthetic approaches including asymmetric methodologies have been developed for epoxide opening with fluoride. To name a few, fluorine-containing reagents such as pyridine·HF [14-15], Et3N·3HF [16], BF3·OEt2 [17], HBF4 [18], KHF2 [19], TBAF/KHF2, TBAF·3H2O/KF [20], KHF2/18-crown-6/chiral Jacobsen chromium complex [21], AgHF2 with Ru catalyst [22], and benzoyl fluoride with a base and HFIP [23] have been reported. In spite of such a plethora of reagents, the stereo- and regioselective transformation of varied functionalized alicyclic frameworks is still a challenge. Bicyclic oxirane-fused ring systems, in particular, are still considered to be complicated substrates for ring opening with fluoride. Accordingly, there is certainly a demand for the development of novel methodologies in view of this type of fluorination.
Results and Discussion
The aim of the current paper is to describe a fluorination protocol based on the oxidative transformation of substituted cyclohexenes through epoxidation, followed by nucleophilic opening of the three-membered O-heterocyclic ring system with fluoride by using Deoxofluor or XtalFluor-E reagents. This is a continuation of our effort to explore the chemical behavior of various epoxy-2-aminocyclohexane- or cyclopentanecarboxylates in selective fluorinations described in a recent paper [24]. In addition to study the anchimeric effect of the functional groups, the synergic influence of simultaneously present carboxylate and N-protected amino moieties on the outcome of fluorination was also investigated. Accordingly, in view of these earlier results, here we describe the opening of some oxiranes, which contain only a single functional group on the cyclohexene skeleton: either a carboxylate or a protected amino group.
Our experimental investigation was started with commercially available racemic ethyl cyclohexane-3-carboxylate (±)-1. First, it was submitted to epoxidation according to a literature procedure [25] furnishing, due to stereochemical factors, epoxycyclohexane esters (±)-2 and (±)-4 in yields of 55% and 29%, respectively. Opening of the epoxide ring in (±)-2 was attempted under various conditions in view of the fluorinating reagent (Deoxofluor, XtalFluor-E), solvent (CH2Cl2, THF or 1,4-dioxane) and temperature. Unfortunately, Deoxofluor did not give any identifiable product. After some attempts, however, we found that the opening of the oxirane ring proceeded with 2 equiv of XtalFluor-E, in the presence of Et3N HF in refluxing dioxane, leading regioselectively through the fluoride attack on C-6 to a fluorine-containing product. The structure of product fluorohydrine (±)-3 was assigned on the basis of 2D NMR data (Scheme 1). Noteworthy, neither of the use of a larger amount of XtalFluor-E reagent, nor Deoxofluor provided the expected hydroxy–fluorine exchange product, only either the unreacted starting ester or decomposed materials were detected.
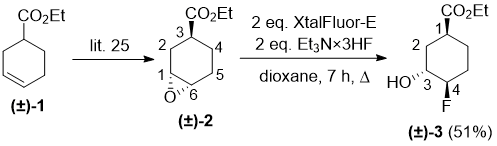
Scheme 1.
The favoured diaxial conformations during the oxirane opening may explain the regioselectivity of the above reaction. The attack of the fluoride ion to the oxirane ring at position C-6 leads to the favoured chair diaxial conformation with the hydroxy and fluorine located in axial positions, affording the 4-fluorinated derivative. The fluoride ion attack to the oxirane ring at position C-1 would generate an unfavoured twisted chair diaxial conformation with the fluorine and hydroxy groups in diaxial orientations; therefore the corresponding 3-fluorinated derivative was not formed (Scheme 2; note: the difference in the numbering of the epoxide system and the fluoro-substituted product!).
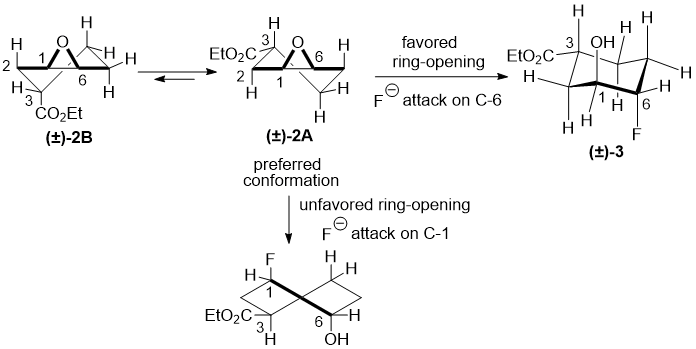
Scheme 2.
Next, epoxide (±)-4 was subjected to fluorination under various experimental conditions. 2 equiv of XtalFluor-E induced oxirane ring opening in the presence of Et3N·HF in refluxing dioxane and furnished regioselectively a fluorinated product, which was identified according to 2D NMR data as compound (±)-5, with the fluorine attached to C-3 on the cyclohexane skeleton.
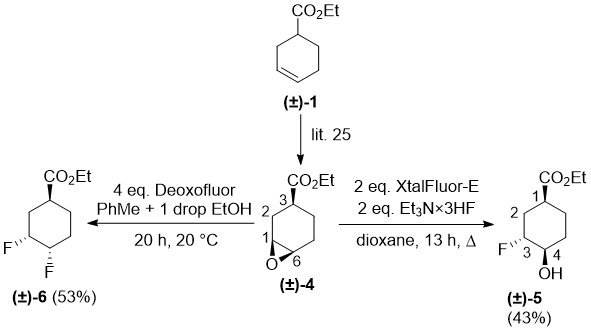
Scheme 3.
Again, the regiosolectivity of this epoxide opening may be explained by a favourable diaxial chair conformation as depicted in Scheme 4.
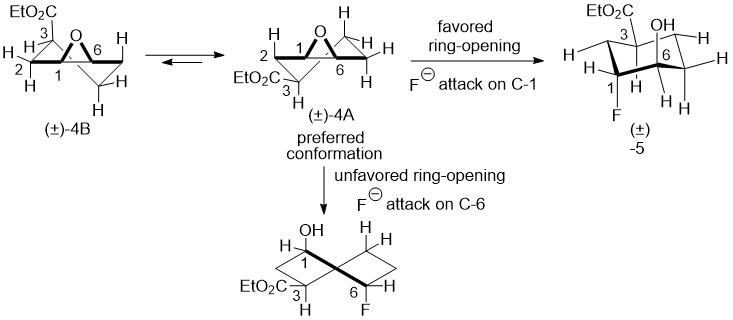
Scheme 4.
Further attempts to exchange the newly created hydroxy group in (±)-5 for fluorine, either by XtalFluor-E or Deoxofluor remained uneffective. Interestingly, contrary to the behaviour of epoxide (±)-2, stereoisomer (±)-4 with 4 equiv of Deoxofluor in the presence of 1 drop of EtOH in toluene resulted in a fluorinated product, identified as (±)-5. It was formed in regioselective oxirane opening and hydroxy–fluorine substitution through configurational inversion (Scheme 3).
In continuation, investigations on amino-substituted cyclohexene were performed. Cbz-Protected cyclohexene (±)-8, derived from commercially available cyclohex-3-enecarboxylic acid (±)-7 [26], was submitted to epoxidation with MCPBA yielding only a single stereoisomer identified as (±)-9 in a cis-diastereoslective manner. In this product, the carbamate group and oxirane ring have the same steric arrangement relative to the six-membered ring. The stereoslectivity of this reaction may be attributed to the hydrogen-bonding directing effect in the transition state of the oxidation [27, 28]. Surprisingly, fluorination of (±)-9 with XtalFluor-E did not afford any identifiable product. In contrast, the reaction with Deoxofluor induced fluorination through regioselective epoxide opening, followed by hydroxy–fluorine interconversion leading to difluorinated derivative (±)-10 as the sole product (Scheme 5). Compare this to a similar process to form (±)-6 shown in Scheme 3.
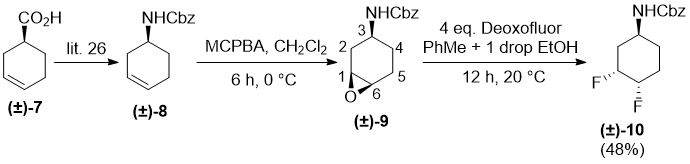
Scheme 5.
Conclusions
Cyclohexene derivatives as model compounds have been studied in an oxidation and fluorination sequence to access highly functionalized fluorine-containing alicyclic scaffolds. The synthetic procedure was based on the epoxidation of the ring olefinic bond followed by ring opening of the resulting functionalized cyclohexane-fused oxiranes with both XtalFluor-E and Deoxofluor as fluorinating reagents.
Experimental
General information:
Chemicals were purchased from Sigma-Aldrich. Solvents were used as received from the suppliers. Melting points were determined with a Kofler apparatus. Elemental analyses were performed with a Perkin–Elmer CHNS-2400 Ser II elemental analyser. Silica gel 60 F254 was purchased from Merck. NMR spectra were acquired at room temperature on a Bruker Avance 400 spectrometer with 11.75 T magnetic field strength (1H frequency 400.13 MHz; 19F frequency 376.50 MHz, 13C frequency 100.76 MHz) in CDCl3 or D6-DMSO solution, using the deuterium signal of the solvent to lock the field. The 1H and 13C chemical shifts are given relative to TMS and 19F to CFCl3 (0.00 ppm).
Ethyl (1S*,3R*,6R*)-7-oxabicyclo[4.1.0]heptane-3-carboxylate and ethyl (1R*,3R*,6S*)-7-oxabicyclo[4.1.0]heptane-3-carboxylate
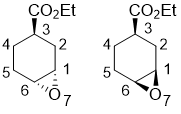
To a solution of ethyl cyclohex-3-enecarboxylate (1.87 g, 12.1 mmol) in CH2Cl2 (75 mL), 3-chloroperbenzoic acid (1.2 equiv) was added. After stirring at room temperature for 2 hours, the reaction mixture was diluted with CH2Cl2 (70 mL), washed with a solution of Na2SO3 (1.3 g in 30 ml saturated aqueous NaHCO3 solution), then washed with 2 × 30 ml saturated NaHCO3 solution. The organic layer was dried (Na2SO4) and concentrated. The product mixture was separated by column chromatography on silica gel (n-hexane/EtOAc). Yields: 55% ethyl (1S*,3R*,6R*)-7-oxabicyclo[4.1.0]heptane-3-carboxylate and 29% ethyl (1R*,3R*,6S*)-7-oxabicyclo[4.1.0]heptane-3-carboxylate (lit. 25).
Benzyl (1S*,3S*,6R*)-7-oxabicyclo[4.1.0]heptan-3-ylcarbamate
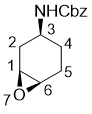
Benzyl cyclohex-3-en-1-ylcarbamate (0.50 g, 2.16 mmol) was dissolved in CH2Cl2 (25 mL), cooled to 0 °C, then 3-chloroperbenzoic acid (1.2 equiv) was added. After stirring for 4 hours, during which the reaction mixture was allowed to warm up to room temperature, the reaction mixture was diluted with CH2Cl2 (25 mL), washed with a solution of Na2SO3 (0.5 g in 25 ml saturated aqueous NaHCO3 solution), then washed with 2 × 25 ml saturated NaHCO3 solution. The organic layer was dried (Na2SO4) and concentrated. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc).
White solid; Yield 75%; Rf = 0.50 (n-hexane/EtOAc 3:2); Mp 56-65 °C. 1H-NMR (400 MHz, D6-DMSO, TMS) δ = 1.18-1.31 (m, 1H, H-4), 1.34-1.43 (m, 1H, H-4), 1.52-1.62 (m, 1H, H-2), 1.75-1.87 (m, 1H, H-5), 1.98-2.07 (m, 1H, H-5), 2.12-2.22 (m, 1H, H‑2), 3.01-3.09 (m, 2H, H-1 and H-6), 3.21-3.28 (m, 1H, H-3), 4.99 (s, 2H, OCH2), 7.08 (d, J = 7.40 Hz, 1H, NH), 7.27-7.40 (m, 5H, CH-Ar). 13C NMR (100 MHz, D6-DMSO, TMS) δ = 24.6, 25.5, 31.0, 46.2, 51.2, 51.8, 66.0, 128.6, 129.2, 138.1, 156.1. MS (ESI, pos) m/z 248 (M + 1). Anal. Calcd. for C14H17NO3: C, 68.00; H, 6.93; N, 5.66. Found: C, 67.69; H, 6.66; N, 5.31.
General procedures for oxirane ring opening with fluoride:
Method A
To a solution of starting epoxide (0.50–0.75 mmol) in anhydrous toluene (10 mL) under an Ar atmosphere, one drop of EtOH and 50% Deoxofluor in toluene (4 equiv) were added and the solution was stirred at 20 °C for the time given in the text. The solution was then diluted with CH2Cl2 (30 mL) and the organic layer was washed with saturated aqueous NaHCO3 solution (2 × 20 mL). The organic layer was dried (Na2SO4) and concentrated. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc).
Method B
To a solution of starting epoxide (0.50–0.75 mmol) in 1,4-dioxane (10 mL) under an Ar atmosphere Et3N·3HF (2 equiv) was added, followed by the addition of XtalFluor-E (1 equiv). The reaction mixture was refluxed for the time given in the text. The solution after cooling was diluted with CH2Cl2 (30 mL) and the organic layer was washed with saturated aqueous NaHCO3 solution (2 × 20 mL). The organic layer was dried (Na2SO4) and concentrated. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc or n‑hexane/acetone).
Ethyl (1S*,3R*,4R*)-4-fluoro-3-hydroxycyclohexanecarboxylate
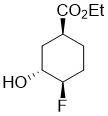
Colorless oil; Yield 51%; Rf = 0.47 (n-hexane/EtOAc 2:1). 1H-NMR (400 MHz, CDCl3, TMS) δ = 1.27 (t, J = 7.12 Hz, 3H, CH3), 1.56-1.83 (m, 3H, H-2, H-5 and H-6), 1.89-2.09 (m, 2H, H-5 and H-6), 2.19-2.31 (m, 1H, H-2), 2.67-2.79 (m, 1H, H‑1), 3.90-4.02 (m, 1H, H-3), 4.16 (q, J = 7.12 Hz, 2H, OCH2), 4.28-4.49 (m, 1H, H-4). 19F NMR (376 MHz, CDCl3) δ = -184.71. 13C NMR (100 MHz, CDCl3, TMS) δ = 14.6, 24.3 and 24.4 (3J = 7.21 Hz), 26.7 and 26.9 (2J = 19.17 Hz), 32.2 and 32.3 (3J = 3.64 Hz), 38.1, 61.0, 69.1 and 69.3 (2J = 22.58 Hz), 92.9 and 94.6 (1J = 172.97 Hz), 174.9. Anal. Calcd. for C9H15FO3: C, 56.83; H, 7.95. Found: C, 56.53; H, 7.69.
Ethyl (1S*,3R*,4R*)-3-fluoro-4-hydroxycyclohexanecarboxylate
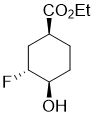
Colorless oil; Yield 43%; Rf = 0.41 (n-hexane/acetone 4:1). 1H-NMR (400 MHz, CDCl3, TMS) δ = 1.27 (t, J = 7.12 Hz, 3H, CH3), 1.51-1.69 (m, 2H, H-5 and H-6), 1.74-1.86 (m, 1H, H-2), 1.86-2.07 (m, 2H, H-5 and H-6), 2.23-2.41 (m, 1H, H-2), 2.70-2.79 (m, 1H, H-1), 3.73-3.84 (m, 1H, H-4), 4.15 (q, J = 7.12 Hz, 2H, OCH2), 4.49-4.70 (m, 1H, H-3). 19F NMR (376 MHz, CDCl3) δ = -185.90. 13C NMR (100 MHz, CDCl3, TMS) δ = 14.6, 24.3, 28.4 and 28.5 (3J = 3.76 Hz), 30.2 and 30.4 (2J = 19.83 Hz), 38. 8 and 38.9 (3J = 7.28 Hz), 61.1, 70.5 and 70.7 (2J = 22.29 Hz), 91.8 and 93.5 (1J = 171.89 Hz), 174.7. Anal. Calcd. for C9H15FO3: C, 56.83; H, 7.95. Found: C, 56.51; H, 8.38.
Ethyl (1S*,3R*,4S*)-3,4-difluorocyclohexanecarboxylate
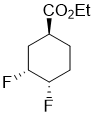
Colorless oil; Yield 53%; Rf = 0.47 (n-hexane/EtOAc 9:1). 1H-NMR (400 MHz, CDCl3, TMS) δ = 1.26 (t, J = 7.14 Hz, 3H, CH3), 1.49-1.64 (m, 1H, H-6), 1.70-1.88 (m, 1H, H-2), 1.88-2.14 (m, 3H, 2 × H-5 and H-6), 2.26-2.39 (m, 1H, H-2), 2.66-2.76 (m, 1H, H-1), 4.14 (q, J = 7.12 Hz, 2H, OCH2), 4.41-4.67 (m, 1H, H-4), 4.84-5.06 (m, 1H, H‑3). 19F NMR (376 MHz, CDCl3) δ = -188.58, -200.35. 13C NMR (100 MHz, CDCl3, TMS) δ = 14.6, 25.4 and 25.5 (3J = 9.91 Hz), 25.7 and 25.7 and 25.9 and 25.9 (2J = 19.96 Hz, 3J = 3.33 Hz), 31.2 and 31.2 and 31.4 and 31.4 (2J = 20.34 Hz, 3J = 5.59 Hz), 37.1 and 37.2 (3J = 2.93 Hz), 61.1, 87.4 and 87.6 and 89.2 and 89.3 (1J = 177.81 Hz, 2J = 17.43 Hz), 89.3 and 89.5 and 91.1 and 91.3 (1J = 181.41 Hz, 2J = 18.18 Hz), 174.8. Anal. Calcd. for C9H14F2O2: C, 56.24; H, 7.34. Found: C, 55.88; H, 6.99.
Benzyl (1S*,3R*,4S*)-3,4-difluorocyclohexyl)carbamate
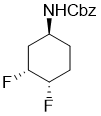
White solid; Yield 48%; Rf = 0.53 (n-hexane/EtOAc 2:1); Mp 102-106 °C. 1H-NMR (400 MHz, D6-DMSO, TMS) δ = 1.27-1.43 (m, 1H, H-6), 1.50-1.71 (m, 1H, H-2), 1.72-1.95 (m, 3H, H-6 and 2 × H-5), 2.01-2.17 (m, 1H, H-2), 3.56-3.72 (m, 1H, H-1), 4.57-4.84 (m, 1H, H-4), 4.85-5.12 (m, 3H, H-3 and OCH2), 7.21-7.44 (m, 6H, CH-Ar and NH). 19F NMR (376 MHz, D6-DMSO) δ = -185.60, -196.57. 13C NMR (100 MHz, D6-DMSO, TMS) δ = 25.3 and 25.5 (2J = 20.04 Hz), 28.6, 34.6 and 34.8 (2J = 19.21 Hz), 45.2, 66.2, 88.7 and 88.9 and 90.4 and 90.6 (1J = 174.10 Hz, 2J = 17.29 Hz), 89.5 and 89.7 and 91.3 and 91.4 (1J = 178.17 Hz, 2J = 17.80 Hz), 128.7, 129.2, 138.0, 156.3. MS (ESI, pos) m/z 270 (M + 1). Anal. Calcd. for C14H17F2NO2: C, 62.44; H, 6.36; N, 5.20. Found: C, 62.10; H, 6.09; N, 4.86.
Acknowledgments
We are grateful to the Hungarian Research Foundation (NKFIH Nos. K 115731 and K 119282) for financial support. The financial support of the GINOP-2.3.2-15-2016-00014 project is also acknowledged. This research was supported by the EU-funded Hungarian grant EFOP-3.6.1-16-2016-00008.
References and Notes
- Wang, P. A. Beilstein J. Org. Chem. 2013, 9, 1677-1695.
- Zhu, Y.; Wang, Q.; Cornwall, R. G.; Shi, Y. Chem. Rev. 2014, 114, 8199-8256.
- Chawla, R.; Singh, A. K.; Yadav, L. D. S. RSC Adv. 2013, 3, 11385-11403.
- Meninno, S.; Lattanzi, A. Chem. Eur. J. 2016, 22, 3632- 3642.
- Kiss, L.; Remete, A. M.; Nonn, M.; Fustero, S.; Sillanpaa, R.; Fülöp, F. Tetrahedron 2016, 72, 781-787 (and references cited therein).
- Zhou, Y.; Wang, Y.; Gu, Z.; Wang, S.; Zhu, W.; Acena, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 442-518.
- Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432-2506.
- O’Hagan D. J. Fluorine Chem. 2010, 131, 1071-1081.
- Kiss, L.; Forró, E.; Fustero, S.; Fülöp, F. Eur. J. Org. Chem. 2011, 4993-5001.
- Nonn, M.; Kiss, L.; Haukka, M.; Fustero, S.; Fülöp, F. A Org. Lett. 2015, 17, 1074-1077 (and references therein).
- Champagne, P. A.; Desroches, J.; Hamel, J. D.; Vandamme, M.; Paquin, J. F. Chem. Rev. 2015, 115, 9073-9174.
- Wu, J. Tetrahedron Lett. 2014, 55, 4289-4294.
- Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem. Int. Ed. 2013, 52, 8214-8264.
- Husstedt, W. S.; Wiehle, S.; Stillig, C.; Bergander, C.; Grimme, S.; Haufe, G. Eur. J. Org. Chem. 2011, 355-363.
- O’Hagan, D. J. Org. Chem. 2012, 77, 3689-3699.
- Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2016, 14, 1117-1123.
- Fraile, J. M.; Mayoral, J. A.; Salvatella, L. J. Org. Chem. 2014, 79, 5993-5999.
- Cresswell, A. J.; Davies, S. G.; Lee, J. A.; Morris, M. J.; Roberts, P. M.; Thomson, J. E. J. Org. Chem. 2012, 77, 7262-7281.
- Kondapi, V. P. K.; Soueidan, O. M.; Hosseini, S. N.; Jabari, N.; West, F. G. Eur. J. Org. Chem. 2016, 1367-1379.
- Yan, N.; Fang, Z.; Liu, Q. Q.; Guo, X. H.; Hu, X. G. Org. Biomol. Chem. 2016, 14, 3469-3475.
- Haufe, G.; Bruns, S.; Runge, M. J. Fluorine Chem. 2001, 112, 55-61.
- Althaus, M.; Togni, A.; Mezzetti, A. J. Fluorine Chem. 2009, 130, 702-707.
- Kalow, J. A.; Doyle, A. G. J. Am. Chem. Soc. 2011, 133, 16001-16012.
- Remete, A. M.; Nonn, M.; Fustero, S.; Fülöp, F.; Kiss, L. Molecules 2016, 21, 1943.
- Goss, R. J. M.; Lanceron, S.; Roy, A. D.; Sprague, S.; Nur-e-Alam, M.; Hughes, D. L.; Wilkinson, B.; Moss, S. J. ChemBioChem 2010, 11, 598-702.
- Ábrahámi, R. A.; Kiss, L.; Fustero, S.; Fülöp, F. Synthesis 2017, 49, 1206–1213.
- Kiss, L.; Remete, A. M.; Nonn, M.; Fustero, S.; Sillanpaa, R.; Fülöp, F. Tetrahedron 2016, 72, 781-787.
- Kiss, L.; Forró, E.; Sillanpää, R.; Fülöp, F. J. Org. Chem. 2007, 72, 8786-8790.
Supporting Information
1H-NMR and 13C-NMR spectra















Recommended for publication by Prof. József Rábai
Fluorine Notes, 2017, 113, 3-4
