Received: May 2017
DOI 10.17677/fn20714807.2017.03.01
Fluorine Notes, 2017, 112, 1-2
Reaction of perfluorinated alkyl-, alkenyl-, and cycloalkenyltetrafluoro--λ5-iodanes RFIF4 with halide anions in non-aqueous solutions
Vadim V. Bardin
N. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry of Siberian Branch of the Russian Academy of Sciences, Acad. Lavrentjev Ave., 9, 630090 Novosibirsk, Russia
e-mail: bardin@nioch.nsc.ru
Abstract: Perfluoroorganic derivatives of iodine(V) RFIF4 (RF = C6F13, (CF3)2CFCF=CF, cyclo-C6F9) react with halide anions X– (X = Cl, Br, I) in CH2Cl2 and/or in CF3CH2CF2CH3 (PFB) to give mainly iodides RFI. In addition, the corresponding chlorides, bromides and RF were formed. The relative contribution of processes depended on the constitution of perfluoroorganyl moiety. The influence of fluoride anions on the reaction route was studied too. Reaction of [Me4N]F with perfluorocyclohexen-1-yltetrafluoro-λ5-iodane led to salt [Me4N][C6F10IF4] while isomer [Me4N][1-cyclo-C6F9IF5] was not detected.
Key words: perfluoroorganyltetrafluoro-λ5-iodanes, reduction, perfluorocarbanions, NMR 19F spectroscopy.
Chemistry of organoiodine(III) is well studied and achievements in this field are presented in monographs and reviews. Properties of organic derivatives of iodine(V) are significantly less studied [1-7]. The same picture is observed in series of their poly- and perfluorinated analogues [8, 9].
In 2005 we published the simple and convenient method of preparation of perfluorinated alkyl-, alkenyl- and cycloalkenyltetrafluoro-λ5-iodanes RFIF4 by the oxidative fluorination of the corresponding iodides or aryltetrafluoro-λ5-iodanes with xenon difluoride in the presence of BF3 [10] (scheme 1).
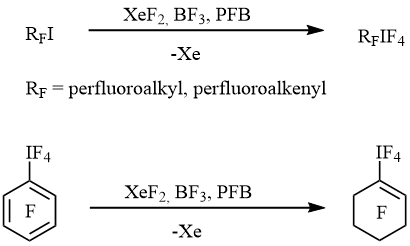
Scheme 1
Chemical properties of these compounds were not investigated. Herein results of interaction of perfluorinated alkyl-, alkenyl- and cycloalkenyltetrafluoro-λ5-iodanes RFIF4 with tetraalkylammonium fluoride, chloride, bromide and iodide in dichloromethane and/or PFB are presented.
An addition of C6F13IF4 (1) in PFB into solution of [Bu4N]Br (in excess) in dichloromethane followed hydrolysis gave a mixture of 1-iodoperfluorohexane (2), 1-bromoperfluorohexane (3) and 1-H-perfluorohexane (4) (molar ratio 5:1:1) in total quantitative yield (scheme 2).
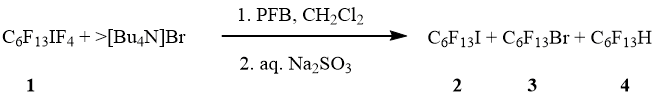
Scheme 2
(Here and below an excess of reagent is marked by ">".)
Reaction of perfluoro-3-methylbuten-1-yltetrafluoro-λ5-iodane (5) (cis : trans = 1 : 9) with [Bu4N]Br in CH2Cl2 led to 1-iodoperfluoro-3-methylbuten-1 (6) and a few trans-1-H-perfluoro-3-methylbuten-1 (7). The ratio cis-6 : trans-6 is the same as in started iodanes 5 (scheme 3).
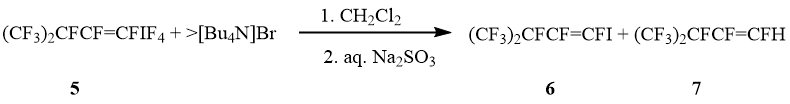
Scheme 3
Under the action of [Bu4N]Br perfluorocyclohexen-1-yltetrafluoro-λ5-iodane (8) underwent to 1-iodoperfluorocyclohexene (9), 1-bromoperfluorocyclohexene (10), and bromoperfluorocyclohexane (11) (molar ratio 3 : 4 : 3) (scheme 4).
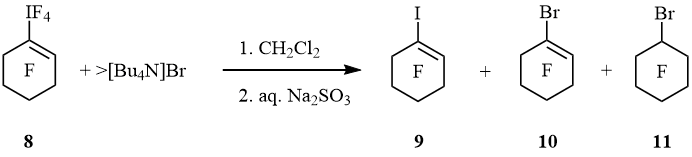
Scheme 4
These results show the processes of replacement of iodine atom by bromine and hydrogen atom that proceeded parallel to the reduction of λ5-iodanes to iodides. Since during the reaction of fluoride anions are formed, we performed a reaction of λ5-iodanes with the [Me4N]F as a "naked" fluoride donor. The action of [Me4N]F on C6F13IF4 in EtCN gave C6F13I (major) and C6F13H (trace). Except they an admixture of unrecognized perfluorohexane derivative, C6F13R, was found (19F NMR). Probably, the reducing agent here is propionitrile. The interaction of iodane 5 with [Me4N]F in MeCN yielded alkene 7 (trace) and substance which is insoluble either in MeCN and DMF (presumably, oligomer). An addition of [Me4N]F to a solution of 8 in CH2Cl2 at –40 °C and sequent warming to 22 °C led to suspension. The liquid phase contained a few 1-H-perfluorocyclohexene (13) but signals of iodane 8 and perfluorocyclohexenes 9 and 10 were not observed (19F NMR). Precipitate was separated, washed with dichloromethane, dried in vacuum and dissolved in anhydrous MeCN. The 19F NMR spectrum displayed resonances of cyclohexene 13, IF5, [IF6]– and salt 12. Signals of isomer [cyclo-C6F9IF5]– that contained hexacoordinated iodine atom were not detected (scheme 5). Up to date, complex [pip][C6H5IF5] produced from hexamethylpiperidinium fluoride, [pip]F, and C6H5IF4 is the only known representative of this family. It was characterized by 19F NMR spectroscopy and X-ray data, but chemical properties were not described [12].
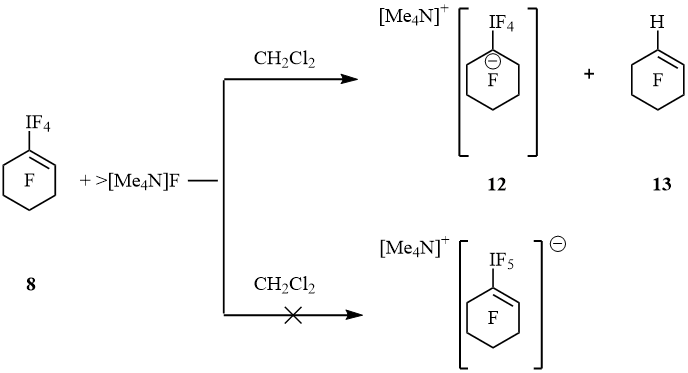
Scheme 5
Spectrum 19F NMR of 12 displayed signals of IF4 group at –13.4 (tt, 4J(F4I, F2, 2) = 20 Hz, 4J(F4I, F6, 6) = 20 Hz, 4F, IF4) and cyclohexyl moiety at –89.3 (m, 4F, F2, 2, 6, 6), –128.9 (m, 4F, F3, 3, 5, 5) and –130.8 (m, 2F, F4, 4) ppm. These values are very close so perfluoro-1-ethyl-cyclohexan-1-ide [–79.5 (CF3), –101.7 (CF2), –87.0 (4F, F2,2,6,6), –129.0 (4F, F3,3,5,5) and –133.0 (2F, F4,4) ppm], that was generated by an addition of fluoride anion to perfluoro-1-ethylcyclohexene [11]. At 22 ºC salt 12 in MeCN converted slowly to cyclohexene 13 and IF5 forming white precipitate. Within 18 h the ratio 12 : 13 was 1 : 1. After treatment of suspension with bromine cyclohexene 10 and additional amount of cyclohexene 13 were obtained (10 : 13 = 1 : 2). It is interesting that the action of [Me4N]F and bromine on 8 in CH2Cl2 resulted in cyclohexenes 9 and 10 (1 : 9) (scheme 6).
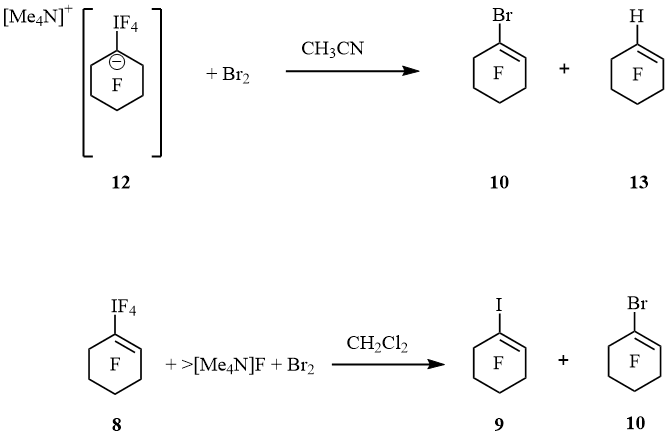
Scheme 6
To complete a picture, reactions of λ5-iodanes 1, 5 and 8 with halide anions in the presence of BF3OMe2 (fluoride anion acceptor) were investigated. Notably that these iodanes resist towards BF3OMe2 as well as the stronger Lewis acid, BF3 [10]. It turned out that in these conditions, iodanes 1 and 5 were transformed into appropriate iodides. Other fluoroorganic products were absent (scheme 7).
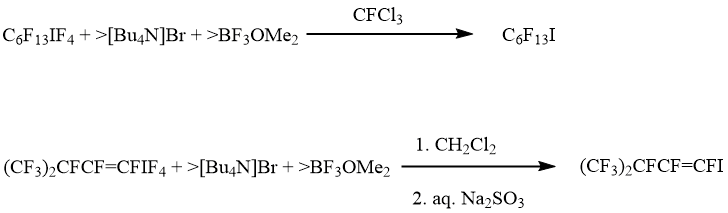
Scheme 7
Alternatively, iodane 8 formed mainly bromocyclohexene 10 while iodocyclohexene 9 was minor product (9 : 10 = 1 : 9). Bromocyclohexane 11 (cf. scheme 4) was not found (scheme 8).
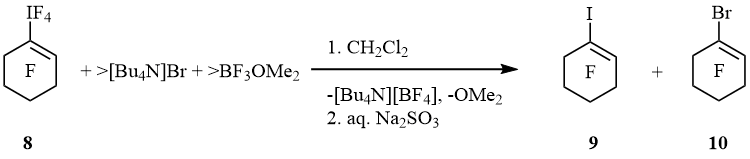
Scheme 8
Iodocyclohexene 9 was the only fluoroorganic product of reaction of 8 with [Bu4N]I and BF3OMe2. When 8 reacted with [Bu4N]Cl and BF3OMe2 that iodocyclohexene was formed together with admixture of 1-chlorononafluorocyclohexene (14) (scheme 9).
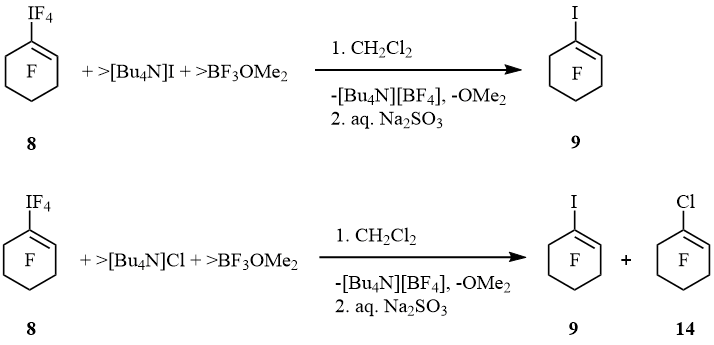
Scheme 9
The resulting picture shows that reactions of λ5-iodanes RFIF4 with halides anions are not the simple reduction of I(V) but proceed several parallel channels. The contribution of each of them determines either by nature of halide and the constitution of perfluoroorganic moiety of iodane.
Experimental part
The 19F NMR spectra were recorded on Bruker AVANCE 300 spectrometer at 282.40 MHz. The chemical shifts are referenced to CCl3F (19F) [with C6F6 as a secondary reference (162.9 ppm)]. The composition of the reaction mixtures and the yields of products were determined by 19F NMR spectroscopy using an internal quantitative standard 1,1,2-trichlorotrifluoroethane or C6F6. Products 2 [13], 3 [14, 15], 4 [16], 9, 10, 13, 14 [17], and 11 [18] were were identified by 19F NMR spectroscopy.
Preparations of C6F13IF4, (CF3)2CFCF=CFIF4, 1-cyclo-C6F9IF4, [10] and [Me4N]F [19], were described in literature. Dichloromethane (Baker), acetonitrile (Riedel-deHaën) were purified by standard procedures. [Bu4N]I (Merck), [Bu4N]Br (Fluka), [Bu4N]Cl (Fluka) were used as supplied. 1,1,1,3,3-Pentafluorobutane (PFB) (Solvay) was stored over molecular sieves 4A. All manipulations were performed in a FEP (block copolymer of tetrafluoroethylene and hexafluoropropylene) equipment under an atmosphere of dry argon.
Reaction of C6F13IF4 1 with [Bu4N]Br
A. Solution of C6F13IF4 (42 mg, 0.08 mmol) in PFB (0.4 ml) was added into stirred solution of [Bu4N]Br (142 mg, 0.44 mmol) in CH2Cl2 (0.7 ml). After 30 min, yellow solution was washed with 5% aq. Na2SO3, with water and dried with MgSO4. The 19F NMR spectrum showed the formation of C6F13I (0.05 mmol), C6F13Br (0.01 mmol) and C6F13H (0.01 mmol).
B. [Bu4N]Br (780 mg, 2.4 mmol) was added in portions into cold (0 C) stirred solution of C6F13IF4 (208 mg, 0.40 mmol) and BF3OMe2 (124 mg, 1.08 mmol) in CFCl3 (4 ml). Immediately red solution was formed. After 10 min the 19F NMR spectrum showed the quantitative formation of 1-iodoperfluorohexane.
Reaction of (CF3)2CFCF=CFIF4 5 with [Bu4N]Br
A. Cold (–15 °C) solution of [Bu4N]Br (317 mg, 0.98 mmol) in CH2Cl2 (1.0 ml) was added in portions into cold (–15 °C) solution of (CF3)2CFCF=CFIF4 (cis : trans = 1 : 9) (104 mg, 0.24 mmol) in CH2Cl2 (0.7 ml) within 5 min. The immediately formed yellow solution was allowed to warm to 22 °C in 30 min, then it was washed with 5% aq. Na2SO3, with water and dried with MgSO4. The 19F NMR spectrum showed the formation of (CF3)2CFCF=CFI (cis : trans = 1 : 9) (0.22 mmol) and trans-(CF3)2CFCF=CFH (0.02 mmol).
B. Cold (–15 °C) solution of [Bu4N]Br (314 mg, 0.97 mmol) in CH2Cl2 (0.7 ml) was added in portions into cold (–15 °C) solution of (CF3)2CFCF=CFIF4 (cis : trans = 1 : 9) (65 mg, 0.15 mmol) and BF3OMe2 (110 mg, 0.96 mmol) in CH2Cl2 (0.5 ml) within 5 min. The immediately formed red solution was allowed to warm to 22°C in 30 min. The 19F NMR spectrum showed resonances of (CF3)2CFCF=CFI (cis : trans = 1 : 9) (0.14 mmol), BF3OMe2 and [Bu4N][BF4]. After washing with 5% aq. Na2SO3, with water and drying with MgSO4. yield of 6 and isomer ratio was not changed.
trans-1-Iodoperfluoro-4-methylbutene-1 (trans-6). 19F NMR (CH2Cl2): = –76.3 (ddd, 5J(F3C, F1) = 5 Hz, 4J(F3C, F2) = 8 Hz, 3J(F3C, F3) = 8 Hz, 6F, F3C), –108.7 (ddsept, 4J(F1, F3) = 40 Hz, 3J(F1, F2) = 151 Hz, 5J(F1, F3C) = 5 Hz,1F, F1), –144.1 (ddsept, 3J(F2, F3) = 15 Hz, 3J(F2, F1) = 151 Hz, 4J(F2, F3C) = 8 Hz, 1F, F2), –187.4 (ddsept, 3J(F3, F2) = 15 Hz, 3J(F3, F1) = 40 Hz, 3J(F3, F3C) = 8 Hz, 1F, F3) (lit. [20] 19F NMR (neat): = –75.4 (6F, F3C), –108.5 (1F, F1), –141.5 (1F, F2), –186.0 (1F, F3); 4J(F2, F3C) = 8.9 Hz, 5J(F1, F3C) = 4.7 Hz, 3J(F3C, F3) = 13.2 Hz, 4J(F1, F3) = 41.4 Hz, 3J(F1, F2) = 148.8 Hz).
cis-1-Iodoperfluoro-4-methylbutene-1 (cis-6). 19F NMR (CH2Cl2): = –75.7 (dd, 3J(F3C, F3) = 9 Hz, 4J(F3C, F2) = 9 Hz, 6F, F3C), –83.5 (dd, 4J(F1, F3) = 6 Hz, 3J(F1, F2) = 10 Hz, 1F, F1), –128.5 (m, 1F, F2), –177.1 (dsept, 4J(F3, F1) = 6 Hz, 3J(F3, F3C) = 9 Hz, 1F, F3).
Reaction of 1-cyclo-C6F9IF4 8 with [Bu4N]Br
A. Solution of 1-cyclo-C6F9IF4 (20 mg, 0.05 mmol) in CH2Cl2 (0.4 ml) was added into stirred solution of [Bu4N]Br (58 mg, 0.18 mmol) in CH2Cl2 (0.2 ml). The obtained solution contained 1-cyclo-C6F9I (0.015 mmol), 1-cyclo-C6F9Br (0.019 mmol) and bromoundecafluorocyclohexane (0.015 mmol) (19F NMR). After washing with 5% aq. Na2SO3, with water and drying with MgSO4. yields were not changed.
B. Solution of 1-cyclo-C6F9IF4 (58 mg, 0.13 mmol) in CH2Cl2 (0.7 ml) was added into stirred solution of [Bu4N]Br (188 mg, 0.58 mmol) and BF3OMe2 (61 mg, 0.54 mmol) in CH2Cl2 (0.5 ml). After 15 min, red solution contained 1-cyclo-C6F9I (0.01 mmol), 1-cyclo-C6F9Br (0.10 mmol) beside [Bu4N][BF4] ((F) = –151.0 ppm) (19F NMR). Solution was washed with 5% aq. Na2SO3, with water and dried with MgSO4. Yields of 9 and 10 were not changed (19F NMR).
Reaction of 1-cyclo-C6F9IF4 8 with [Bu4N]Cl and BF3OMe2
Solution of 1-cyclo-C6F9IF4 (58 mg, 0.13 mmol) in CH2Cl2 (0.7 ml) was added into stirred solution of [Bu4N]Cl (180 mg, 0.65 mmol) and BF3OMe2 (61 mg, 0.54 mmol) in CH2Cl2 (0.5 ml). After 15 min, yellow solution contained 1-iodononafluorocyclohexene (0.09 mmol) and 1-chlorononafluorocyclohexene (0.01 mmol) beside [Bu4N][BF4] (19F NMR).
Reaction of 1-cyclo-C6F9IF4 8 with [Bu4N]I and BF3OMe2
Solid [Bu4N]I (500 mg, 1.35 mmol) was added in portions into stirred solution of 1-cyclo-C6F9IF4 (152 mg, 0.34 mmol) and BF3OMe2 (124 mg, 1.08 mmol) in CH2Cl2 (5 ml). Immediately iodine evolved and deep violet solution showed the presence of 1-cyclo-C6F9I and [BF4]- (19F NMR). Solution was washed with 5% aq. Na2SO3, with water and dried with MgSO4. The 19F NMR spectrum contained resonances of 1-cyclo-C6F9I 9 (quantitative yield) and [BF4]-.
Reaction of 1-cyclo-C6F9IF4 8 with [Me4N]F
Cold (–0 °C) solution of 1-cyclo-C6F9IF4 (31 mg, 0.07 mmol) in CH2Cl2 (0.4 ml) was added into cold (–40 °C) stirred solution of [Me4N]F (18 mg, 0.19 mmol) in CH2Cl2 (0.2 ml). The reaction mixture was warmed to 22 °C over 20 min to form suspension. After 1 h white precipitate was separated by centrifugation from brownish mother liquor which contained only trace of fluoroorganic compounds (19F NMR). Precipitate was separated by centrifugation, washed with dichloromethane and dried in vacuum to give white solid (32 mg). Freshly prepared solution in anhydrous MeCN contained 12, 13, [Me4N][IF6] (singlet at 11.7 ppm [21]), and IF5 (ratio 12:42:7:39) (19F NMR). After 18 h at 22 ºC the ratio became 20:21:7:53.
Reaction of [Me4N][C6F10IF4] 12 with bromine in MeCN
Cold (–15 °C) solution of bromine (20 mg, 0.125 mmol) in MeCN (0.08 ml) was added into cold (–15°C) solution of 12 in MeCN (see above). Salt 12 quantitatively converted to cyclohexenes 10 and 13 (3 : 7) (19F NMR).
Reaction of 1-cyclo-C6F9IF4 8 with bromine and [Me4N]F
Cold (–20 °C) solution of [Me4N]F (22 mg, 0.23 mmol) in CH2Cl2 (0.4 ml) was added into cold (–20 °C) stirred solution of 1-cyclo-C6F9IF4 (31 mg, 0.07 mmol) and bromine (32 mg, 0.20 mmol) in CH2Cl2 (0.7 ml). Immediately white suspension formed. The reaction mixture was warmed to 22 °C over 10 min, and precipitate was separated by centrifugation. Yellow mother liquor contained cyclohexenes 10 (0.06 mmol) and 9 (trace). Solution of solid in CH3CN contained [Me4N][IF6] (19F NMR).
Reaction of C6F13IF4 1 with [Me4N]F
Cold (0 °C) solution of [Me4N]F (18 mg, 0.19 mmol) in EtCN (0.5 ml) was added into cold (–55 °C) stirred solution of 1(66 mg, 0.12 mmol) in EtCN (0.6 ml). The reaction mixture was warmed to 22 °C over 6 h. Yellow solution contained C6F13I (0.09 mmol), C6F13H (trace) and unknown product C6F13R (R ≠ IF2, IF4) (19F NMR).
Acknowledgements
Author gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (436 RUS 17/12/04) and the Fonds der Chemischen Industrie.
References
- V.V. Zhdankin // Science of Synthesis 31a (2007) Chapter 31.4.1.
- Hypervalent Iodine Chemistry. Modern Developments in Organic Synthesis // Eds. T. Wirth, G.F. Koser / Topics in Current Chemistry 224 (2003) 1.
- Akiba K.-Y. Chemistry of Hypervalent Compounds.–New York: Wiley-VCH, 1999.
- Varvoglis A. Hypervalent Iodine in Organic Synthesis.–London: Academic Press, 1997.
- Varvoglis A. The Organic Chemistry of polycoordinated Iodine.– Weinheim: VCH, 1992.
- E.D. Matveeva, M.V. Proskurnina, N.S. Zefirov // Heteroatom Chem. 17 (2006) 595-617.
- N.S. Pirkuliev, V.K. Brel, N.S. Zefirov // Russ. Chem. Rev. 69 (2000) 105-120.
- H.-J. Frohn, M.E. Hirschberg, A. Wenda, V.V. Bardin // J. Fluorine Chem. 129 (2008) 459-473.
- V.V. Bardin // Fluorine Notes 80 (2012); http://notes.fluorine1.ru/
- H.-J. Frohn, V.V. Bardin // J. Fluorine Chem. 126 (2005) 1036-1043.
- N.I. Delyagina, S.M. Igumnov, V.F. Snegirev, I.L. Knunyants // Bull. Acad. Sci. USSR, D. Chem. Sci. (1981) 1836-1840.
- S. Hoyer, K. Seppelt // J. Fluorine Chem. 125 (2004) 989-996.
- A.S. Otazaghine //J. Fluorine Chem. 126 (2005) 1009-1016.
- B.-N. Huang, A. Haas, M. Lieb //J. Fluorine Chem. 36 (1987) 49-62.
- E. Signe, H. Blancou, A. Commeyras //J. Fluorine Chem. 70 (1995) 197-200.
- T. Hudlicky, R. Fan, J.W. Reed, D.R. Carver, M. Hudlicky, E.I. Eger // J. Fluorine Chem. 59 (1992) 9-14.
- S.F. Campbell, A.G. Hudson, E.F. Mooney, A.E. Pedler, R. Stephens, K.N. Wood // Spectrochim. Acta 23A (1967) 2119-2125.
- J.W. Emsley // Mol. Phys. 9 (1965) 381-394.
- K.O. Christe, W.W. Wilson, R.D. Wilson, R. Bau, J. Feng // J. Am. Chem. Soc. 112 (1990) 7619-7625.
- V.F. Cherstkov, M.V. Galakhov, S.R. Sterlin, L.S. German, I.L. Knunyants // Bull. Acad. Sci. USSR, D. Chem. Sci., (1986) 104-108.
- K.O. Christe, W.W. Wilson // Inorg. Chem. 28 (1989) 3275-3277.
Recommended for publication by Prof. S. M. Igumnov
Fluorine Notes, 2017, 112, 1-2
