Received: april 2017
DOI 10.17677/fn20714807.2017.02.01
Fluorine Notes, 2017, 111, 1-2
The Peculiarities of Electrochemical Behavior of ω-Bromopolyoxaperfluorocarboxylic Acids
V.A. Grinberg1, A.A. Tyutyunov2, 3, N.D. Kagramanov2, N.A. Mayorova1, E.I. Maevskii4, S.R. Sterlin2
1A.N. Frumkin Institute of Physical Chemistry and Electrochemistry, RAS, Leninskii Prosp.
31, 119071, Moscow, Russia
2A.N. Nesmeyanov Institute of Organoelement Compounds,
RAS, ul. Vavilova 28, V-334, GSP-1, 119991 Moscow, Russia
3NPO PiM-INVEST
LLC, ul. Vavilova 28, 119991 Moscow, Russia
4Institute of Theoretical and Experimental
Biophysics, RAS, Institutskaya ul. 3, 142290, Pushchino, Moscow Region, Russia
e-mail: sterlins@yandex.ru
Annotation: It is shown that electrochemical behavior of ω-bromopolyoxafluorocarboxylic acids of the general formula BrCF2CF2O[CF(CF3)CF2O]nCF(CF3)CO2H (n = 0÷3) under the conditions of anodic oxidation (Kolbe electrosynthesis) differs dramatically depending on the length of their main chain.
Key words: ω-bromopolyoxafluorocarboxylic acids; Kolbe electrosynthesis; α, ω-dibromopolyoxaperfluoroalkanes.
Kolbe electrosynthesis is one of the preparative techniques most in demand in electrochemistry. This reaction is used for preparation of dimers of aliphatic radicals, generated at carboxylate anions electrooxidation. Primarily it refers to the dimers of fluorinated radicals, inclusive of functionalized ones, stable under electrosynthesis conditions (e.g. preparation of α, ω-bis(fluorosulfonyl)perfluoroalkanes via electrooxidation of carboxylate anions of -fluorosulfonylperfluorocarboxylic acids [1]).
It was shown as early as in 1956 that in the course of electrooxidation of ω-halogeno-carboxylic acids of the general formula Hal(CH2)nCO2H (where Hal = F, Cl, Br, J) the formation of the corresponding Kolbe dimers was observed provided that the main chain of the acids reached a definite length (Hal = F, n = 4; Hal = Cl, n = 2; Hal=Br, n = 10) [2]. The authors remarked that upon increasing the value of n up to 10 the yield of Kolbe dimers steadily increased from 39 to 82% for various halogeno-carboxylic acids. Contrary to that, all ω-iodocarboxylic acids (containing from 2 to 11 carbon atoms in the molecule) underwent decomposition accompanied by iodine evolution without formation of dimer products.
The goal of the given study is a synthesis of α, ω-dibromopolyoxaperfluoroalkanes possessing, just as other fluorocarbons, a high oxygen-dissolving capacity, decreased volatility, increased lipophilicity in comparison with perfluoroalkanes and capable to form emulsions stable both during prolonged storage at above-zero temperatures and under the conditions of sterilization in autoclave at 120oC. Such compounds can be used for preparation of oxygenating emulsions – therapeutic agents of external application (creams, ointments and liniments), as well as oxygenating components of culture media in biotechnological processes.
In order to solve this important problem we studied electrochemical behavior of ω-bromopolyoxaperfluorocarboxylic acids of the general formula BrCF2CF2O[CF(CF3)CF2O]nCF(CF3)CO2H (n = 0÷3) [1a (n = 0); 1b (n = 1); 1c (n = 2); 1d (n = 3)] and found out that it changes cardinally depending on the length of the acid’s main chain. As it was shown, the anodic oxidation of the carboxylate anions of acids 1a-b resulted in a formation of multicomponent mixture of products, in which, however, the expected Kolbe dimers haven’t been detected.
At the same time, the electrolysis of partially neutralized acid 1b in the presence of CF3CO2H led to the formation of Kolbe cross-dimer 3 (50% yield) (Scheme 1), that indicates unambiguously at the generation of radical 2 as a result of decarboxylation of ω-bromofluoroacyloxy radical – primary intermediate of the carboxylate anion 1b oxidation.
Scheme 1
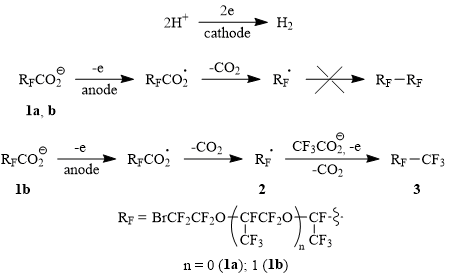
In contrast to their lower homologues electrolysis of partially neutralized acids 1c-d gives both Kolbe homo- and cross-dimers 4-6 in good yields (at Scheme 2 1c’ and 1d’ are carboxylate anions of the corresponding acids). The formation of dimers 4-6 is controlled readily by 19F NMR spectra: a signal of fluorine atom in a fragment CF(CF3)СO2H (δ: -132, m) disappears, and new signals of fluorine atoms in fragments of dimerized products CF(CF3)-CF(CF3) (δ: -143, m) appear instead. Herein the current yield of the dimers grows up while the electrolyte temperature rises up to 50÷55oC that is most probably connected with deglobulization of the starting acids:
Scheme 2
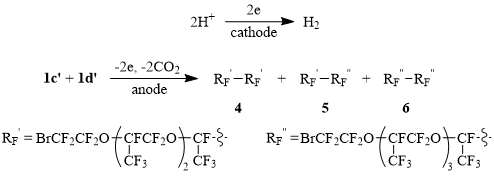
The cathode reaction in Schemes 1 and 2 is the proton reduction resulting in hydrogen evolution.
Thus, we ascertained that the acids 1a-b, just like linear -halogeno-carboxylic acids [2], exhibit abnormal electrochemical behavior and do not form dimerization products of the corresponding alkyl radicals under Kolbe electrosynthesis conditions.
It should be taken into consideration that the branched polyoxaperfluorocarboxylic acids RCF2CF2O[CF(CF3)CF2O]nCF(CF3)CO2H (R = CF3, FSO2; n = 1÷3) – close structural analogues of acids 1a-b – dimerize smoothly under anodic oxidation conditions [3-5]. Also it’s worth noting that the adsorption energy of 3-oxa-2-trifluoromethylperfluorocaproate anion is higher than that of perfluorovalerate anion of similar molecular mass: as it was shown earlier, the anodic oxidation of equimolar mixture of C3F7OCF(CF3)CO2H and C4F9CO2H in the presence of butadiene gave a mixture of Kolbe homo- and cross-dimers R’-R’ : R’-R’’ : R’’-R’’ (R’ = C3F7OCF(CF3); R’’ = C4F9) in a ratio 2,2 : 3,3 : 1, and that indicated at a preferable displacement of butadiene from anode by 3-oxa-2-trifluoromethylperfluorocaproate anion [6]. The authors’ assumption that enhanced adsorption of this carboxylate anion is connected with the presence of oxygen atom in the main chain of the molecule, acting as an additional nucleophilic centre and forming a donor-acceptor bond with the anode material, is in agreement with the evidence of the enhanced persistence and, correspondingly, lowered reactivity of perfluorinated α-alkoxyalkyl radicals as compared with their perfluoroalkyl analogues [7].
In its turn, the enhanced persistence of α-alkoxyalkyl radicals can be explained by formation of mesomeric forms (Scheme 3) due to electron-donating ability of unshared electron pair at oxygen atom in the α-position towards paramagnetic centre [8]:
Scheme 3
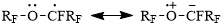
It is clear that the above mentioned pertains equally to acids 1. But acids 1, in addition to ethereal oxygen atoms, contain a bromine atom in the ω-position, its covalent radius being considerably larger than that of oxygen atom (1.14 and 0.66 Å, correspondingly [9]).
One can assume that polarization of bromine atom takes place under the electrolysis conditions (anode potential > 2.1 V vs SCE), that leads to the formation of another donor-acceptor bond which, in its turn, enhances yet more the energy of adsorption of both carboxylate anions and radicals, generated in the course of electrooxidation, at the anode.
The immobilization of radicals at the anode surface favors their further oxidation to carbocations (Hofer-Möest reaction) that in aq. MeCN medium should lead to the formation of unstable perfluorinated esters of CF3CO2H. Their degradation and subsequent hydrolysis should form a mixture of CF3CO2H and bromodifluoroacetic acid (in the case of acid 1a) and CF3CO2H and acid 1a (in the case of acid 1b) (Scheme 4):
Scheme 4
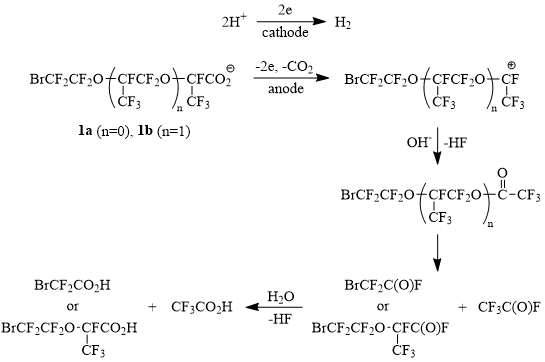
When passing from acids 1a-b to their higher homologues - acids 1c-d, the donor-acceptor bonding of the oxygen and bromine atoms with anode material weakens under the influence of steric shielding, the latter growing symbatically with the increasing number of side trifluoromethyl substituents (competitive advantage of steric factors is most probably favored by globulization of radicals due to the presence of “hinged” oxygen atoms). As a result the generated bromopolyoxafluoroalkyl radicals are capable to go into the solution bulk forming dimerized products (cf. with data [10]).
Thereby it is shown that the anodic oxidation of acids 1c-d is a convenient method for preparation of low-volatile and chemically inert organofluorine liquids, that can be used as oxygenating additives to therapeutic agents of external application and as components of culture media in microbiology.
Experimental
19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 282 MHz. Chemical shifts are given in ppm vs. CFCl3. Downfield shifts are positive. Mass spectra are recorded using a VG FYFLITICAL 7070E (70 eV) and a Finnigan Polaris/GCQ Plus (70 eV) mass spectrometers (Trace GC ultra). The acids 1a-b were obtained by saponification of the corresponding acyl fluorides of general formula BrCF2CF2O[CF(CF3)CF2O]nCF(CF3)COF, n = 0, 1 [11].
Preparation of acids 1c-d.
Water (5 ml, 280 mmol) was added to a mixture of BrCF2CF2O[CF(CF3)CF2O]2CF(CF3)COF (30 g, 44 mmol) and 10 ml of hexane. After 30 min of vigorous stirring 5 ml of H2SO4 was added to the reaction mixture, the lower organic layer was separated and distilled under reduced pressure collecting the fraction boiling within the range 90-150оC/1Torr.
The redistillation gave 24.7 g (82%) of 11-bromo-3,6,9-trioxaperfluoro-2,5,8-trimethylundecanoic acid (1c) (a mixture of diastereomers), b.p. 133÷135оC/17 Torr. Calc. for C11BrHF20O5, %: С 19.61; H 0.15; F 56.43; Br 11.89. Found, %: С 20.00; H 0.20; F 56.37; Br 11.36.
The spectrum 19F NMR of acid 1c:
1 2 3 4 5 6 7 8 9 10
BrCF2CF2OCF(CF3)CF2OCF(CF3)CF2OCF(CF3)CO2H
-70.9 (2F1); -79.2÷-85.3 (15F2+4+5+7+8+10); -132.1 (1F9); -146.1÷-146.6 (2F3+6)
14-Bromo-3,6,9,12-tetraoxaperfluoro-2,5,8,11-tetramethyltetradecanoic acid (1d) (a mixture of diastereomers), b.p. 118÷122оC/0.1 Torr was obtained likewise by saponification of BrCF2CF2O[CF(CF3)CF2O]3CF(CF3)COF. Calc. for C14BrHF26O6, %: С 20.02; H 0.12; F 58.87; Br 9.53. Found, %: С 20.31; H 0.15; F 59.02; Br 8.49.
The spectrum 19F NMR of acid 1d:
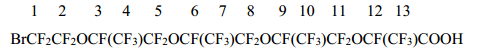
-70.97 (2F1); -79.2÷-85.3 (20F2+4+5+7+8+10+11+13); -132.1 (1F12); -146.1÷-146.6 (3F3+6+9).
Preparation of cross-dimer 3 from acid 1b and CF3CO2H.
A mixture of acid 1b (10 g, 19.7 mmol), CF3CO2H (3.42 g, 30 mmol), KOH (0.224 g, 4 mmol), 29 ml MeCN and 1 ml H2O was electrolyzed at a Pt/10% Ir (15 cm2) anode using a stainless steel cathode (2 cm2) in a one-compartment cell at 20oC (current 1.0A, current density 64.4 mA.cm-2, initial voltage 24 V, theoretical charge for consumption of 49.7 mmol of acids -1.33 A.h). The final voltage after passing 1.99 A.h was 18 V. Then the electrolyte was washed with water and aq. K2CO3 solution, the organic layer was separated and distilled to give 5.1 g (yield 49% by substance and 26% by current) of cross-dimer 3 [BrCF2CF2OCF(CF3)CF2OCF(CF3)2], b.p. 129÷131oC. Calc. for C8BrF17O2, %: C 18.08; F 60.83. Found, %: C 17.98; F 60.74.
Mass spectrum of 3 (m/z, reference): 511[M-F]+; 451[M-Br]+; 395[C6F12BrO]+; 335[C6F13O]+; 179[C2F4Br]+; 147[C3F5O]+; 100[C2F4]; 69[CF3]+(100%); 47[CFO]+; 31[CF]+.
Preparation of dibromide 4.
A mixture of acid 1c (9.8 g, 14 mmol), KOH (0.122 g, 2.2 mmol), 29 ml MeCN and 1 ml H2O was electrolyzed at a Pt/10% Ir anode (15cm2) using a stainless steel cathode (2 cm2) in a one-compartment cell at 40÷50oC (current 0.8 A, current density 51.6 mA.cm-2, initial voltage 24 V, theoretical charge for consumption of 11.8 mmol of acid - 0.32A.h). The final voltage after passing 0.37 A.h was 40 V. Then the electrolyte was washed with water and aq. K2CO3 solution, the organic layer was separated and distilled to give 7.1 g (yield 80% by substance and 72% by current) of 1,20-dibromo-3,6,9,12,15,18-hexaoxoperfluoro-4,7,10,11,14,17-hexamethyleicozane (4) (diastereomers mixture), b.p. 117÷119oC/1 Torr. Calc. for C20Br2F40O6, %: C 19.11; F 60.51, Br 12.74. Found, %: C 19.09; F 60.43, Br 12.38.
The spectrum 19F NMR of dibromide 4:

-71.1+-72.0 (2F1+2F20); -80.4÷-86.1 (a group of signals 30F2+4+5+7+8+10+12+13+15+16+18+19); -143.2 (1F9+1F11); -147÷-147.5 (4F3 +6+14+17).
Mass spectrum of 4 (m/z, reference): 727[C12BrF24O3]+; 705[C12BrF22O4]+; 667[C12F25O3]+; 611[C12BrF22O3]+; 551[C10F21O2]+; 529[C10F19O3]+; 511[C8BrF16O2]+; 395[C6BrF12O2]+; 385[C8BrF16O2]+; 345[C5BrF10O]+; 335[C6F13O]+; 313[C6F11O2]+; 285[C5F11O]+; 229[C3BrF6]+; 219[C4F9]+; 179[C2BrF4]+; 147[C3F5O]+; 131[C3F5]+; 100[C2F4]+; 69[CF3]+(100%).
Preparation of dibromide 6.
Dibromide 6 was obtained from 10.0 g (11.9 mmol) of acid 1d and 0.1 g (1.7 mmol) of KOH by the same procedure as the dibromide 4. The treatment of electrolyte after electrolysis (as described above) afforded 6.8 g (yield 72% by substance and 56% by current) of 1,26-dibromo-3,6,9,12,15,18,21,24-octaoxoperfluoro-4,7,10,13,14,17,20,23-octamethylhexacozane (6) (diastereomers mixture), b.p. 183÷187oC/1 Torr. Calc. for C26Br2F52O8, %: C 19.66; F 62.21. Found, %: C 19.91; F 62.37.
The spectrum 19F NMR of dibromide 6:
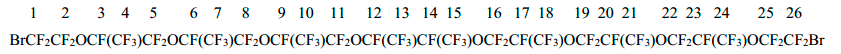
-71.8 (2F1+2F26); -80.6÷-86.3 (a group of signals 40F2+4+5+7+8+10+11+13+15+16+18+19+21+22+24+25); -143.0 (2F12+14); -146.5÷-147.5 (6F3 +6+9+17+20+23).
Mass spectrum of 6 (m/z, reference): 961[C18F35O5]+; 912[C15BrF31O4]+; 871[C14BrF30O4]+; 843[C14BrF28O4]+; 811[C15F29O5]+; 679[C13F25O3]+; 645[C12F23O4]+; 607[C12F21O4]+; 501[C9F19O2]+; 451[C8F17O2]+; 420[C7F16O2]+; 385[C7F15O]+; 335[C6F13O]+; 313[C6F11O2]+; 285[C5F11O]+; 219[C4F9]+; 179[C2BrF4]+; 147[C3F5O]+; 131[C3F5]+; 97[C2F3O]+; 69[CF3]+(100%).
Preparation of dibromides 4, 5, 6.
A mixture of acid 1c (4.9 g, 7 mmol), acid 1d (5.9 g, 7 mmol), KOH (0.1 g, 1.7 mmol), 29 ml MeCN and 1 ml H2O was electrolyzed at a Pt/10% Ir anode (15 cm2) using a stainless steel cathode (2 cm2) in a one-compartment cell at 50oC (current 0.8 A, current density 51.6 mA.cm-2, initial voltage 28 V, theoretical charge for consumption of 12.3 mmol of acids - 0.32 A.h). The final voltage after passing 0.37 A.h was 55 V. The treatment of electrolyte after electrolysis (as described above) afforded 7.5 g (yield 72% by substance and 56% by current) of the dibromides 4, 5, 6 mixture (4 : 5 : 6 = 1 : 2 : 1 according to GLC data).
The analytical sample of 1,23-dibromo-3,6,9,12,15,18,21-heptaoxaperfluoro-4,7,10,11,14,17,20-heptamethyltricosane (5) (a mixture of diastereomers), b.p. 150÷153oC/1 Torr was isolated by further rectification. Calc. for C23Br2F46O7, %: C 19.41; F 61.46. Found, %: С 19.53; F 61.37.
The spectrum 19F NMR of dibromide 5:
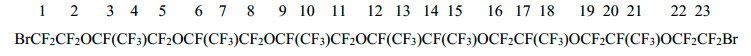
-71.8 (2F1+2F23); -80.6÷-86.3 (a group of signals 35F2+4+5+7+8+10+11+13+15+16+18+19+21+22); -143.0 (2F12+14); -146.5÷-147.5 (5F3 +6+9+17+20).
Mass-spectrum of 5 (m/z, reference): 912[C15BrF31O4]+; 871[C14BrF30O4]+; 843[C14BrF28O4]+; 811[C15F29O5]+; 795[C15F29O4]+; 727[C12BrF24O3]+; 667[C12F25O3]+; 611[C12BrF22O3]+; 551[C10F21O2]+; 511[C8BrF16O2]+; 451[C8F17O2]+; 385[C8BrF16O2]+; 335[C6F13O]+; 313[C6F11O2]+; 285[C5F11O]+; 219[C4F9]+; 179[C2BrF4]+; 147[C3F5O]+; 131[C3F5]+; 100[C2F4]+; 69[CF3]+(100%).
References
- V.F. Cherstkov, V.A. Grinberg, S.R. Sterlin, Yu.B. Vassiliev, L.S. German, Izv.Akad.Nauk.Ser.Khim., 1990, 10, 2448.
- F.L.M. Pattison, J.B. Stothers, R.G. Woolford, J.Am.Chem.Soc., 1956, 78, 2255.
- G. Jager, H. Millauer, US patent №5,468,352 (1995).
- M.A. Efremova, G.I. Kaurova, N.B. Lesnevskaia, A.A Ludikainen, V.A. Matalin, N.V. Peganova, N.V. Puzanova, Izv.SPbGTI (TU), 2011, 37(11), 102.
- N.V. Peganova, G.I. Kaurova, N.B. Lesnevskaia et al., Zhurn.Prikl.Khim., 2009, 82(12), 1976.
- N.A. Maiorova, N.D. Kagramanov, V.A. Grinberg, S.R. Sterlin, Russ.J.Electrochem., 2013, 49(2), 181.
- V.A. Grinberg, S.R. Sterlin, Izv.Akad.Nauk,Ser.Khim., 2005, 8, 1884.
- D.C. Nonhebel, J.C. Walton, Free-Radical Chemistry. University Press. Cambridge. 1974, P. 62.
- F. Cotton, J. Wilkinson, Contemporary inorganic chemistry. Part I, Mir Publ., Moscow, 1969, P. 127.
- V.F. Cherstkov, V.A. Grinberg, S.R. Sterlin, Yu.B. Vassiliev, L.S. German, E.I. Mysov, Izv.Akad.Nauk,Ser.Khim., 1991, 5, 1134.
- S.M. Igumnov, S.R. Sterlin, A.A. Tyutyunov, Z.A. Mikhailova, RF patent № 2497801 (2013).
Recommended for publication by PhD A.A. Tyutyunov
Fluorine Notes, 2017, 111, 1-2
