Received: November, 2013
Fluorine Notes, 2013, 91, 5-6
Preparative method for obtaining of higher fluoroalkyl silanes
V. E. Boykoab, A.A. Tyutyunovab, V. L. Donab, S. M. Igoumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, V-334, GSP-1, 119991 Moscow, Russia
b ZAO NPO "P&M-Invest", 119334, Moscow, Leninsky avenue 47
e-mail: boykii@mail.ru
Abstract: A preparative method for obtaining of silanes Rf Si(CH3)3 with length Rf more than C4was developed. Also silanes from Methyl 2-(2-bromo-1,1,2,2-tetrafluoroethoxy)-2,3,3,3-tetrafluoropropionate (n=1) and its oligomer (n=2) were obtained for the first time.
Keywords: (fluoroalkyl)trimethylsilanes, (perfluorohexyl)trimethylsilane, (perfluorooctyl)trimethylsilane, methyl 2,3,3,3-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-(trimethylsilyl)ethoxy)propionate.
(Fluoroalkyl)trimethylsilanes are unique reagents for induction of fluoroalkyl arrangement in different classes of organic and organoelement compounds. [1-3] considering its chemical behavior they are similar to Grignard reagent. At the same time they are convenient to work with, as unlike Grignard reagents, they can be stored for a long time without being decomposed. More over fluorinated alkyl silanes are less sensitive to the presence of substrate of different functional groups in molecule.
Since Ruppert for the first time obtained (trifluoromethyl)trimethylsilane in 1984, which afterwards was named after him, through reaction of trimethyliodide with trimethylchlorosilane in the presence of tris(diethylamino)phospine [4], and its trifluoromethylation under conditions of nucleophilic catalisis, appeared a lot of works, dedicated to the methods for obtaining of Ruppert reagent itself and other fluorinated silanes. [1,5-9].
(Trifluoromethyl)trimethylsilane, also brom-, and chlorodifluoromethyl-, and (pentafluoroethyl)trimethylsilane are obtained through interaction between fluoroalkylhalides with trimethylchlorosilanes in the presence of metallic Al, Mg or Li. [10-15]. Also silanes with chain length C1-C4 were obtained through interaction of perfluorohalides in diglyme with trimethylchlorosilane and tetrakis(dimethylamino)ethylene [16] with output 55 – 81 %. There are also information about obtaining of (perfluorohexyl)trimethylsilane through the same method, the output is approximately 40 % [17].
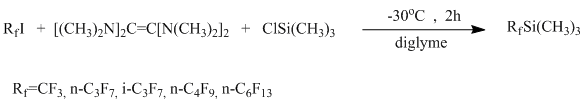
Moreover there are several examples of obtaining (perfluoroalkyl)trimethylsilanes with the chain length >4.
So (perfluorohexyl)trimethylsilane was obtained through interaction of perfluorohexyl iodide with trimethylchlorosilane in the presence of samarium iodide with output 68 % [18] .
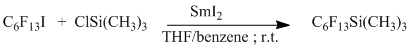
A process of (perfluooroctyl)trimethylsilane through interaction of perfluorooctyl magnesium iodide or bromide with trimethylchlorosilane in THF with outputs from 33 to 77 % is described. [19-22].
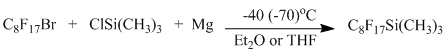
Previously we offered a method of synthesis of (perfluoroalkyl)trimethylsilanes through interaction of perfluoroalkylhalides with silanization agents (Me3SiOC(O)CF3, Me3SiOSO2Me) in the presence of zinc-copper couple under conditions of Barbier reaction [23].
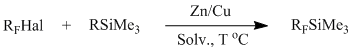
R = Cl, CF3CO2; CH3SO3
RF = CF3;
C2F5; nC3F7; iC3F7;
C4F9; CF2CO2Me; CF2CO2Et; CF2CF2OCF=CF2.
Therefore we obtained different trimethylsilanes, including perfluoroalkyl and perfluoroalkenyl silanes, however all (perfluoroalkyl)trimethylsilanes that we obtained had the lenth of carbon chain till C4 [21]. These silanes, after formation, flake away from reaction mixture with upper layer, so they are withdrawn from interaction. By the attempt to obtain silanes with longer length of Rf , as C4 , we observed in 19F NMR Spectra signals in the area, characteristic for RfH. As for the signals in the area, characterisctic for RfSi , we observed it only in the noise level, which make us assume that obtainig of silanes with longer length Rf , as C4, is not posible.
However, in the course of further studies, we found out that, the more time passes from the time of ending the process to the time of finishing of isolation of silane from reaction mixture, the more content of perfluoroalkyl hydrides (RfH) is observed.
The initial data of 19F spectroscopy can be explained with the fact that, from the moment of sampling to the moment of taking spectra was enough time for protodesilylation of appeared product. One can avoid it, by treatment of reaction mixture with solvent of chlorohydric or sulphuric acid right after carrying the process.
Therefore from corresponding bromides (1a) and (1b), we obtained (perfluorohexyl)trimethylsilane (2a) and (perfluorooctyl)trimethylsilane (2b) with outputs 89 and 77 % correspondingly.
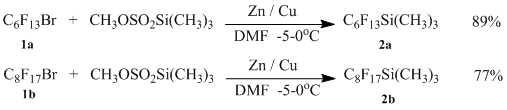
In the same way, we obtained silanes (4a) and (4b) from methyl 2-(2-bromo-1,1,2,2-tetrafluoroethoxy)-2,3,3,3-tetrafluoropropionate (n=1) (3a) and its oligomer (n=2) (3b).
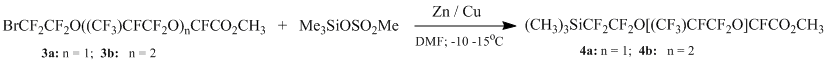
This reaction is of special interest, due to the possibility of dealkoksilylation of ethers (3a) and (3b) in the presence of zink.
Summarizing all the above information, we developed a preparative method for obtaining of silanes with length of Rf mote then C4. There were also for the first time obtained silanes from methyl 2-(2-bromo-1,1,2,2-tetrafluoroethoxy)-2,3,3,3-tetrafluoropropionate (3a) (n=1) and methyl 2,3,3,3-tetrafluoro-2-(1,1,2,3,3,3-hexafluoro-2-(1,1,2,2-tetrafluoro-2-ethoxy)propoxy)propionate ) (3b).
Experimental
NMR 19F spectra are taken on spectrometer “Bruker AM300” by 282 MHz, ESTD is CDCl3 or D2O. The chemical shifts 19F are given per million relative CFCl3. 1H per million relative TMS. Spectral data for silanes (2a) and (2b) are presented in the literature (24, 20).
(Perfluorohexyl)trimethylsilane (2a)
In a three-neck flask of volume 250 ml to 100 ml of dimethyl formamide while stirring 13 g zink, 0,5 g CuCl and 1g trimethylchlorosilane are added, then allowed for 30 minutes, after that the reaction mixture is cooled to 0 °C and 20.2 g of trimethylsilyl ether of methanesulfonic acid are dropwise added, then mixture is cooled to -5 °C and 40 g of perfluorohexylbromide are dropwise added .Further it is allowed for one more hour by -5 °C, after that it is poured in stirred mixture of H2SO4 with ice of 20 % concentration. The bottom layer is isolated, dried with Magnesium sulfate, filtered and rerun. 35 g of perfluorohexyltrimethylsilane in the form of colorless liquid are obtained. The boiling point is 147 °C. The yield is 89 %.
NMR 19F :-83,26(t,3F,CF3), -120,44(c,2F), -123,50(c,2F, CF2),-124,51 (c,2F, CF2),-128,06(c,2F ,CF2),-130,05 (c, 2F, CF2Si); 1H NMR:0,5 (c,9H,)
(Perfluorooctyl)trimethylsilane (2b)
In a three-neck flask of volume 250 ml to 100 ml of DMF while stirring 10,5 g zink, 0,4 g CuCl and 1g trimethylchlorosilane are added, then allowed for 30 minutes, after that the reaction mixture is cooled to 0 °C and 16.2 g of trimethylsilyl ether of methanesulfonic acid are dropwise added, then mixture is cooled to -5 °C and 40 g of perfluorooctylbromide are dropwise added . Further it is allowed for one more hour by -5 °C, after that it is poured in stirred mixture of H2SO4 with ice of 20 % concentration. The bottom layer is isolated. It is rerun with water vapour, dried with magnesium sulfate, filtered and rerun in vacuum. 30 g of trimethyl(perfluorooctyl)silane in the form of colorless liquid are obtained. The boiling point is 75 °C /20 mm Hg. The yield is 77 %. NMR Spectra 19F -82,76 (t.,3F,CF3), -129,67(s.2F,CF2Si),-127,67(s.2F,CF3CF2),-123,09(m.,8F,(CF2)4)), -120( m.,2F,CF2CF2Si).
1H NMR:0,7(s,9H,Si(CH3)3)).
Methyl 2,3,3,3-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-(trifluoromethylsilyl)ethoxy)propionate (4a)
In a three-neck flask of volume 250 ml to 150 ml of DMF while stirring 11 g zink, 0,5 g CuCl and 1g trimethylchlorosilane are added, then allowed for 30 minutes, after that the reaction mixture is cooled to 0 °C and 17 g of trimethylsilyl ether of methanesulfonic acid are dropwise added, then mixture is cooled to -10 (-15) °C and 30 g of methyl 2-(2-bromo-1,1,2,2-tetrafluoroethoxy)-2,3,3,3-tetrafluoropropionate are dropwise added. Further it is allowed for one more hour by -10 (-15) °C, after that it is poured in stirred mixture of H2SO4 with ice of 20 % concentration. The bottom layer is isolated, dried with magnesium sulfate, filtered and rerun in vacuum. 15 g of methyl 2,3,3,3-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-(trifluoromethylsilyl)ethoxy)propionate in the form of colorless liquid is obtained. The yield is 51 %. The boiling point is 34 °C/ 20 mm Hg.
| Spectra NMR 19F | : |
-83,89 (d,3F,CF43), from -81,64 to -90,19 (m, 2F,CF22O), -132,23(s, 2F,CF12Si),-132,99(d,1F,CF3);
1H NMR:0,5 (s,9H,Si(CH3)3)),4,2(s,3H,OCH3).
Methyl 2,3,3,3-tetrafluoro-2-(1,1,2,3,3,3-hexafluoro-2-(1,1,2,2-tetrafluoro-2-(trimethylsilyl)-ethoxy)propoxy)propionate (4b)
In a three-neck flask of volume 250 ml to 150 ml of DMF while stirring 13 g zink, 0,5 g CuCl and 1,1 g trimethylchlorosilane are added, then allowed for 30 minutes, after that the reaction mixture is cooled to 0 °C and 20 g of trimethylsilyl ether of methanesulfonic acid are dropwise added, then mixture is cooled to -10 (-15) °C and 52 g of methyl 2,3,3,3-tetrafluoro-2-(1,1,2,3,3,3-hexafluoro-2-(1,1,2,2-tetrafluoro-2-(trimethylsilyl)ethoxy)-propoxy)propionate are dropwise added. Further it is allowed for one more hour by -10 (-15) °C, after that it is poured in stirred mixture of H2SO4 with ice of 20 % concentration. The bottom layer is isolated, dried with magnesium sulfate, filtered and rerun in vacuum. 36 g of methyl 2,3,3,3-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-(trifluoromethylsilyl)ethoxy)propionate in the form of colorless liquid is obtained. The yield is 71 %. The boiling point is 52 °C/ 5 mm Hg.
| Spectra NMR 19F | 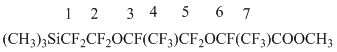 |
: |
-82,03(d,3F,CF63), -84,49(t.,3F,CF43), from -80,13 till -87,45 (m 4F,CF2,52O), -131,97(m.,2F,CF12Si), -133,39, -147,20 (m 2F,CF3+7), 1H NMR: 0,4 (s.9H,Si(CH3)3)), 4,1(s.3H,OCH3).
References
- G.K.S. Prakash, A.K. Yudin, Chem.Rev., 1997, 97, 757-786.
- J.-A. Ma, D. Cahard, Chem.Rev., 2004, 104, 6119-6146 (2008, 108, PR1-PR43).
- K. Uneyama, J.Fluor.Chem., 2008, 129, 550-576.
- I. Ruppert, K. Schlich, W. Volbach, Tetrahed.Lett., 1984, 25, 2195-2198.
- R.P. Singh, J.M. Shreeve, Tetrahedron, 2000, 56, 7613-7632.
- S. Roy, B.T. Gregg, G.W. Gribble, V.-D. Le, S. Roy, Tetrahedron, 2011, 67, 2161-2195.
- A.D. Dilman, V.V. Levin, Eur.J.Org.Chem., 2011, 831-841.
- O.A. Tomashenko, V.V. Grushin, Chem.Rev., 2011, 111, 4475-4521.
- E.J. Cho, S.L. Buchwald, Org.Lett., 2011, 13, 6552-6555.
- J. Grobe, J. Hegge, ZAAC, 2008, 634, 1975-1990.
- J. Grobe, J. Hegge. Synlett, 1995, 641-642.
- K.I. Petko, S.Y. Kot, L.M. Yagupolskii, J.Fluor.Chem., 2008, 129, 301-306.
- S.M. Igoumnov, V.A. Vyazkov, A.V. Prokhorov, RF Patent 2399624, 2009.
- S.M. Igoumnov, E.V. Igoumnova. Sintezy ftororganicheskikh soedinenii (Syntheses of Organofluorine Compounds), Part 1, 2nd ed., ZAO NPO P&M-Invest, Moscow, 2010.
- S. Utsumi, T. Katagiri, K. Uneyama, Tetrahedron, 2012, 68, 580-583.
- V.A. Petrov, Tetrahedron Letters , 2001 v.42, , 3267-3269.
- Murphy-Jolly, B. Makeba, Lewis, L. Lesley, Caffyn, J.M. Andrew, Chemical Communication (Cambridge, U.K.), 2005, 35, 4479-4480
- M. Yoshida, D. Suzuki, M Iyoda, J. Chem. Soc., Perkin Trans. 1, 1997, 5, 643-648.
- E.I. Du Pont de Nemours and Co., US Patent 5093512 1992.
- C.F. Smith, E.J. Soloski, C. Tamborski, J.Fluor.Chem., 1974, 4, 35-45.
- L.G. Vaughan, W.A. Sheppard, J. American Chem. Soc., 1969, 91, 6151-6156.
- G.J.Chen, L.S.Chen, K.S.Eapen, W.E.Ward, J.Fluor.Chem., 1974, 69, 61-66.
- S.M. Igoumnov, V.K. Men'shikov, V.E. Boyko, A.A. Tyutyunov, S.R. Sterlin //Fluorine notes, 2012, 6(85), URL: /public/2012/6_2012/letters/letter4.html
- Nagaki, Aiichiro; Tokuoka, Shinya; Yamada, Shigeyuki; Tomida, Yutaka; Oshiro, Kojun; et al., Organic and Biomolecular Chemistry, 2011 , vol. 9, p. 7559 – 7563.
Recommended for publication by Prof. S. Igoumnov
Fluorine Notes, 2013, 91, 5-6
