Received: ноябрь 2016
Fluorine Notes, 2016, 109, 1-2
Исследование реакционной способности трифторметансульфонилбромида по отношению к ненасыщенным соединениям углеводородного ряда
A.A. Тютюновab, Л.Ф. Ибрагимоваa, Н.Д. Каграмановa, С.Р. Стерлинa, С.М. Игумновab
aФедеральное государственное бюджетное учреждение науки Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук, 119991, ГСП-1, Москва, В-334, ул. Вавилова, д. 28
bЗАО НПО “ПиМ-Инвест”, 119991, Москва, ул. Вавилова, д. 28
e-mail: tuytuynov@rambler.ru
Аннотация: Изучено взаимодействие трифторметансульфонилбромида с непредельными соединениями углеводородного ряда в условиях фотохимического, термического и ион-радикального инициирования.
Ключевые слова: Трифторметансульфонилбромид, трифторметилирование.
Алифатические соединения, содержащие терминальную трифторметильную группу, общей формулы CF3(CH2)nX (I), где n = 2-10, X = OH, Br, I, NH2, SH, CHO и т.п., являются востребованными промежуточными продуктами органического синтеза и могут использоваться для получения лекарственных препаратов [1-6], агрохимических средств [7-8], поверхностно-активных веществ [9], жидких кристаллов [10] и т.д. Кроме того, карбинолы I (X = OH, n = 2-4) представляют самостоятельный практический интерес и могут применяться в качестве компонентов смазок [11], чистящих композиций [12], элюентов [13] и экстрагентов [14]. Эти спирты характеризуются коротким периодом полураспада в атмосфере и низкими значениями потенциала глобального потепления (GWP), что позволяет в ряде случаев использовать их в качестве заменителей фреонов [15].
Основными путями синтеза линейных ω-трифторметилсодержащих алифатических производных является как присоединение полигалоалканов (CCl4, CCl3Br) к терминальным олефинам [16] с последующим фторированием аддуктов [8, 17], так и непосредственное введение CF3-группы в углеводородную цепь с использованием реагентов, представленных ниже (схема 1, таблица 1) [18-26].
Схема 1
Таблица 1. Трифторметилсодержащие реагенты и условия их реакций с непредельными соединениями.
CF3-реагент |
Условия |
CF3I Tbp = -22°C |
200°C [27]; 80°C, AIBN [28]; UV-light [27, 29]; Na2S2O4, NaHCO3 [30]; MLn catalyst [20]; Photoredox catalyst [21] |
CF3Br Tbp = -59°C |
Zn, CuI, Ultrasound [31]; Zn, Cp2TiCl2, Ultrasound [31]; 200°C, Pd(PPh3)4 [32]; Aluminum chlorofluoride, 50°C [33] |
CF3SO2Cl Tbp = 32°C |
90÷145°C, ROOR [34]; 125°C, Ru(PPh3)3Cl2 [35-36]; UV-light [24]; Photoredox catalyst [37-41] |
CF3SO2Na |
tBuOOH, Cu salt [42-44]; AgNO3, K2S2O8 [45]; Photoredox catalyst [46-47] |
CF3SO2Br Tbp = 60°C |
20÷65°C [48-49]; sunlight [показано для RFSO2Br [50-51]; 55÷95°C, ROOR [48, 51-52] |
CF3SiMe3 Tbp = 55°C |
CuTc, PhI(OAc)2, K2CO3 [53]; AgNO3, PhI(OAc)2, AcONa [54] |
|
MLn catalyst [22, 24] |
|
TMSN3, [Cu(CH3CN)4]PF6 [55]; CuBr2, SOX2 [56]; MLn catalyst [22, 26, 57] |
CF3CCl3 Tbp = 46°C |
Fe, P(OEt)3, 120°C [58]; UV-light [59] |
CF3CH2I Tbp = 55°C |
Na2S2O4, NaHCO3 [60]; iPr2NEt, fac-Ir(ppy)3, visible light [61] |
CF3CH2SO2Cl Tbp = 140°C |
K2HPO4, fac-Ir(ppy)3, visible light [62]; |
Среди препаратов, используемых для прямого трифторметилирования непредельных соединений (таблица 1), особый интерес представляет трифторметансульфонилбромид – CF3SO2Br (1) как наиболее удобный с препаративной точки зрения трифторметилирующий агент. В то же время имеющиеся в литературе примеры применения 1 в синтезе фторсодержащих соединений носят фрагментарный характер [24, 48-49, 52], что и побудило нас предпринять систематическое изучение реакции трифторметилирования ненасыщенных соединений углеводородного ряда с использованием сульфонилбромида 1, гладко образующегося бромированием трифторметилсульфината калия, получаемого in situ сульфодиоксидированием (трифторметил)триметилсилана (2) [63], или коммерчески доступного трифторметилсульфината натрия (3) [48].
Схема 2
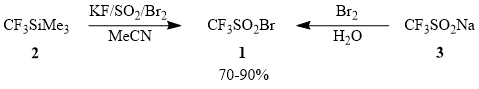
При работе с трифторметансульфонилбромидом (1) следует учитывать, что это соединение, перегоняющееся без разложения (т.кип. 58÷60°С) и способное храниться в темноте длительное время, весьма фоточувствительно и под действием солнечного света претерпевает десульфодиоксидирование с образованием CF3Br. Аналогичный процесс наблюдается при нагревании 1 выше 100°С.
Схема 3
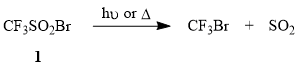
Ранее было показано, что 1 присоединяется к электронодонорным олефинам, например, алифатическим терминальным алкенам, циклогексену, аллилацетату при 20÷65°С, причем реакция сопровождается элиминированием SO2 [48-49]. Однако, не смотря на простоту метода, следует учитывать, что при масштабировании радикальных процессов реакция может выходить из-под контроля и приводить к взрыву (как, например, при присоединении CCl4 к этилену при катализе ПБ [64]).
Нами установлено, что присоединение CF3SO2Br (1) к аллилтрифторацетамиду (4a) и аллилацетату (4b) в запаянных ампулах на солнечном свету (при навесках 4a-b 2-1 моля) осуществляется гладко и приводит к образованию аддуктов 5a-b с выходами 70-90%, тогда как присоединение 1 к гомоаллилацетату (4c) в аналогичных условиях удалось осуществить лишь при загрузке 3-4 ммоля 4c. Более масштабные опыты – с использованием 200-250 ммолей 4c – неизбежно заканчивались взрывом ампулы.
Схема 4
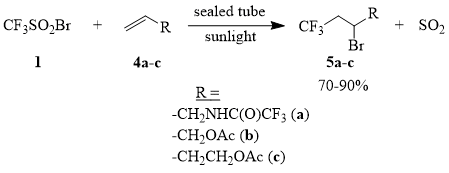
Присоединение 1 к 4a-b в условиях термического инициирования, осуществляется при температурах 70÷100°С. Так, при нагревании 1 и 4a (мольное соотношение 1:1; 100°С/2,5 часа) выход 5a составляет около 50%. Следует отметить, что проведение реакции 1 с 4b в гексане (мольное соотношение 1:1; 65°С/2 часа) в открытой системе привело к образованию 5b с выходом лишь 20%, что значительно меньше выхода 5b по данным работы [48].
Учитывая, что в условиях реакции скорость десульфодиоксидирования 1 существенно возрастает, приводя к снижению выходов целевых продуктов, то термически инициируемое присоединение сульфонилбромида 1 к олефинам 4a-b в открытой системе вряд ли представляет практический интерес.
Известно, что инициирование радикального присоединения полигалоалканов к олефинам эффективно осуществляется различными окислительно-восстановительными системами, например, на основе солей металлов переменной валентности, таких как Cu, Fe, Mn, Ce и др. [65]. Ранее было показано, что галогениды меди являются наиболее активными катализаторами присоединения сульфонилхлоридов и сульфонилбромидов к ряду олефинов и ацетиленов [66-68]. Действительно оказалось, что металлическая медь эффективно инициирует реакцию присоединения 1 к 4a-d в растворе ацетонитрила уже при комнатной температуре. Данный процесс гладко масштабируется и может использоваться для синтеза трифторметилированых продуктов 5a-d.
Схема 5
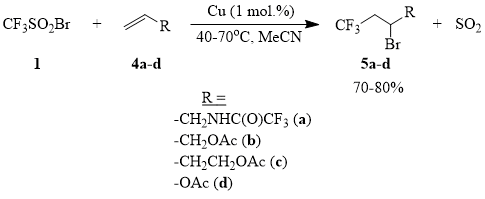
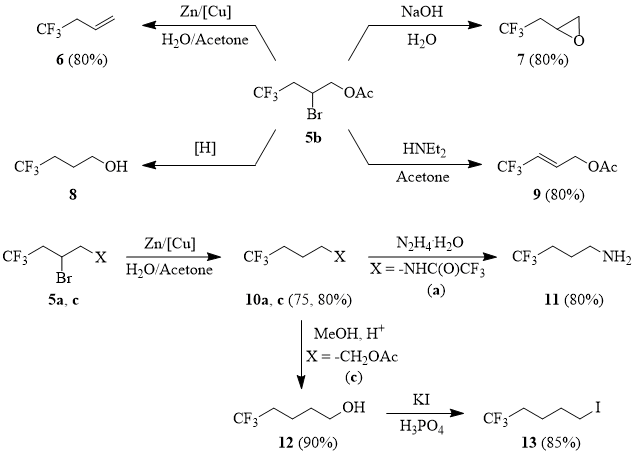
Соединения 5a-d являются ценными полупродуктами, которые могут использоваться для синтеза целого ряда ω-трифторметилсодержащих линейных алифатических производных [69].
Схема 6
Известно, что радикальное присоединение к интернальным олефинам затруднено по сравнению с аналогичными реакциями с терминальными олефинами, что связано со стерическим экранированием винильного атома углерода [70-71]. Неудивительно, что присоединение сульфонилбромида 1 к бутену 14 (запаянная ампула, инсоляция) осуществляется достаточно вяло и сопровождается побочными процессами – образованием CF3Br в результате десульфодиоксидирования 1 и трифторметилсульфиновой кислоты за счет отрыва аллильного атома водорода CF3SO2-радикалом (схема 7, таблица 2). Та же реакция в среде MeCN при инициировании порошкообразной медью привела к образованию аддукта 15 с препаративным выходом, что позволило использовать его в качестве исходного соединения для получения трифторизопрена 17 (схема 8).
Схема 7
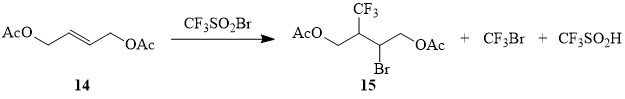
Таблица 2. Реакция CF3SO2Br (1) c 1,4-диацетокси-2-бутеном (14)a.
Условия |
Соотношение продуктов реакции, % |
||
15 |
CF3Br |
CF3SO2H |
|
солнечный свет, 3 дняb |
51 |
34 |
7 |
Cu (2 mol.%), MeCN, rt, 2 дня |
81c |
8 |
10 |
a Соотношение 1:14 = 1:1.
b Запаянная ампула, конверсия 1 93%, загрузка 1 ммоль.
c Препаративный выход 70%
(смесь стереоизомеров).
Схема 8
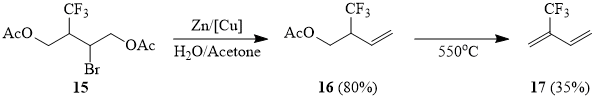
Олефины с электронодефицитной двойной связью – метилакрилат (18a) и акрилонитрил (18b) – также присоединяют сульфонилбромид 1 в присутствии меди в ацетонитриле, однако выходы аддуктов 19a-b ожидаемо ниже по сравнению с выше приведенными примерами. Следует отметить, что теломер-гомологи 19a-b при этом не образуются. В условиях фотохимического инициирования 1 присоединяется к метилакрилату, образуя аддукт 19a с низким выходом, однако при этом наблюдается образование теломер-гомологов 19a (ср. [51]). В этих же условиях вовлечь в реакцию олефин 18b не удается.
Схема 9
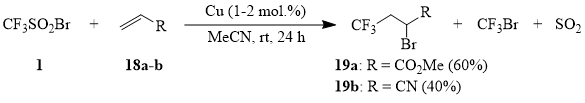
Неожиданно оказалось, что акролеин (20) реагирует с 1 с образованием сульфона 21, причем 2-бром-4,4,4-трифторбутаналь – ожидаемый продукт присоединения CF3-радикала – в продуктах реакции обнаружен не был.
Схема 10
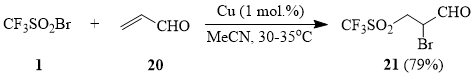
Исследования реакции 1 со стиролом (22) показали, что в этом случае основным продуктом реакции является сульфон 24, о чем ранее не сообщалось [48].
Схема 11
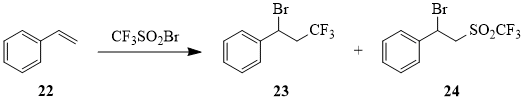
Таблица 3. Реакция CF3SO2Br (1) cо стиролом (22)a.
Условия |
Соотношение продуктов, % |
|
23 |
24 |
|
90°C, Ph(COO)2, hexane |
64b |
- |
солнечный свет, hexane |
34 |
65 |
Cu (1 mol.%), MeCN, 25-30°C |
25 |
75c |
a Соотношение 1:22 = 1:1.
b По данным работы [48].
c При выделении продуктов реакции перегонкой в вакууме сульфон 24 подвергается дегидробромированию (на 50%) с образованием PhCH=CHSO2CF3 (25).
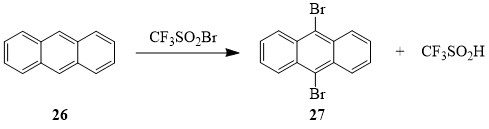
Продукты реакции сульфонилбромида 1 с антраценом (26) – 9,10-дибромантрацен (27) и трифторметилсульфиновая кислота – указывают на то, что в этом случае 1 выступил исключительно в роли бромирующего агента; продуктов заместительного или аддитивного трифторметилирования антрацена обнаружено не было.
Схема 12
Таким образом, показано, что трифторметансульфонилбромид (1) при взаимодействии с реагентами различной природы может быть источником трифторметильной или трифторметансульфонильной группы или выступать в роли бромирующего агента. В то же время установлено, что 1 способен вступать в аддитивные реакции с достаточно широким кругом непредельных соединений углеводородного ряда в условиях фотохимического и ион-радикального катализа, что является удобным препаративным методом синтеза различных классов органических соединений, содержащих трифторметильную группу.
Экспериментальная часть
ЯМР 1H, 19F спектры записаны на спектрометре “Bruker AVANCE-300” при 300 и 282 MHz, соответственно, внешний стандарт CDCl3. Химические сдвиги для 1H спектров приведены относительно остаточного сигнала растворителя (δ 7,26) и даются в м.д. относительно ТМС. Химические сдвиги спектров 19F приведены в м.д. относительно CFCl3. Слабопольные сдвиги имеют положительное значение. Масс-спектры записаны на масс-спектрометре Finnigan Polaris Q (Trace GC ultra).
Трифторметансульфонилбромид (1).
A: Синтез из CF3SiMe3 (2).
Процесс потенциально опасен и может сопровождаться разогревом и выбросом реакционной смеси.
В 6-ти литровую колбу, снабженную механической мешалкой, термометром, капельной воронкой и обратным холодильником, соединенным со склянкой Тищенко с H2SO4(конц.), наливают 2,5 л ацетонитрила (содержание воды 0,04-0,05%). Затем при перемешивании и температуре 15÷20°С присыпают 280 г (4,82 моль) KF и добавляют раствор 720 г (5,06 моль) (трифторметил)триметилсилана (2) и 340 г (5,3 моль) SO2 в 1 л ацетонитрила.
Реакционную смесь перемешивают при 15÷20°С/3-6 часов (возможен разогрев реакционной смеси, температура должна контролироваться внешним охлаждением). В процессе реакции реакционная смесь окрашивается в светло-бурый цвет. Если в течение этого времени реакционная смесь остается практически бесцветной, то перемешивание прекращают и реакционную смесь оставляют стоять на ночь. Затем перемешивание реакционной смеси возобновляют, контролируя её температуру в интервале 20÷30°С. По завершении реакции реакционная смесь приобретает светло-бурую окраску.
Далее реакционную смесь медленно нагревают до температуры 80÷95°С и отгоняют легкокипящие компоненты смеси. Перегонку осуществляют до достижения температуры дистиллята 80°С.
Реакционную смесь охлаждают до 0÷5°С и при перемешивании добавляют по каплям 779 г (4,87 моль) брома. Затем смесь перемешивают в течение 15-30 минут, выливают в полуторакратный объем ледяной воды, нижний слой отделяют, промывают 4 раза равным объемом ледяной воды, к нему при перемешивании добавляют 30 г P2O5 и перегоняют.
Получают 720 г (выход 70%) 1, чистотой 85-90% с примесью FSiMe3 и ацетонитрила. Т.кип. 55÷65°С. ЯМР 19F δ: -78 (с, CF3).
B: Синтез из CF3SO2Na (3) [48].
В 6-ти литровую колбу, снабженную механической мешалкой, термометром, капельной воронкой и обратным холодильником помещают 3 л воды, при перемешивании присыпают 1100 г (4,58 моль) коммерческого трифторметилсульфината натрия (3) (чистота 65 масс.%, примеси NaBr, Na2SO4, Na3PO4), смесь перемешивают в течение 10-15 минут и при температуре 5÷10°С добавляют по каплям 740 г (4,63 моль) брома, до появления устойчивой бромной окраски реакционной смеси.
Нижний слой отделяют, промывают полуторакратным объемом ледяной воды, добавляют к нему при перемешивании 30 г P2O5 и перегоняют.
Получают 878 г (выход 90%) трифторметансульфонилбромида (1), с незначительной примесью брома, для удаления которой к 1 добавляют 0,5-1 масс.% децена-1 и продукт перегоняют, получают бесцветный 1 чистотой 99%. Т.кип. 58÷60°С.
N-(2-бром-4,4,4-трифторбутил)-2,2,2-трифторацетамид (5a).
Реакции в запаянных ампулах потенциально опасны и могут сопровождаться взрывом.
Смесь 590 г (2,77 моль) трифторметансульфонилбромида (1) и 424 г (2,77 моль) аллилтрифторацетамида (4a) запаивают в стеклянную ампулу из молибденового стекла объемом 1 л (диаметром 50 мм и длиной 500 мм) и выдерживают на солнечном свету в течение 1-4 дней (в зависимости от интенсивности солнечного света) на подоконнике. Ампулу охлаждают сухим льдом, вскрывают, вносят кипелки и медленно нагревают до 25÷30°С с такой скоростью, чтобы кипение SO2 было не слишком бурным. Полученный продукт очищают ректификацией в вакууме 0,5-1 Торр, при ректификации в вакууме 15 Торр увеличиваются потери продукта за счет его частичного разложения.
Получают 586-670 г (выход 70-80%) 5a, чистотой 96%. Т.кип. 130°С/15 Торр.
ЯМР 1H δ: 2,7 (м, 2H, CF3CH2), 3,7 (м, 2H, CH2NH), 4,2 (м, 1H, СHBr), 8 (уш.c, 1H, NH); ЯМР 19F δ: -78 (c, 3F, C(O)CF3), -66 (т, 3F, 3JHF = 8 Гц, CF3CH2).
2-Бром-4,4,4-трифторбутилацетат (5b).
Смесь 42,6 г (0,2 моль) трифторметансульфонилбромида (1) и 20 г (0,2 моль) аллилацетата (4b) запаивают в стеклянную ампулу из молибденового стекла объемом 50 мл (диаметром 23 мм и длиной 135 мм) и выдерживают на солнечном свету в течение 1-3 дней (в зависимости от интенсивности солнечного света) на подоконнике. Ампулу охлаждают сухим льдом, вскрывают, вносят кипелки и медленно нагревают до 25÷30°С, отгоняя SO2 с умеренной скоростью. Полученный продукт очищают ректификацией в вакууме 10 Торр.
Получают 39,8 г (выход 80%) 5b, чистотой 98%. Т.кип. 73÷76°С/10 Торр.
ЯМР 1H δ: 2 (с, 3H, CH3), 2,6-2,9 (уш.м, 2H, CF3CH2), 4,2 (м, 3H, CH2O+CHBr); ЯМР 19F δ: -65 (т, 3F, 3JHF = 8 Гц, CF3).
Реакции 1 с алкенами 4a-d в присутствии меди (общая методика).
К раствору (2 моль) олефина 4a-d в 400 мл ацетонитрила добавляют 1,27 г (0,02 моль) порошка меди и при перемешивании и температуре 40÷70°С прибавляют по каплям 426 г (2 моль) трифторметансульфонилбромида (1). После добавления половинного количества 1 начинается газовыделение SO2. Смесь перемешивают в течение 15-60 минут при 70°С, отгоняют растворитель и остаток перегоняют в вакууме 10-0,5 Торр.
Соединения 5b и 5c получают при 65÷70°С, а 5a и 5d при 40÷45°С.
3-Бром-5,5,5-трифторпентилацетат (5c).
Выход 80%. Т.кип. 80-84°С/0,5 Торр.
ЯМР 1H δ: 1,9 (с, 3H, CH3), 2,0-2,3 (уш.м, 2H, CH2СН2О), 2,8 (м, 2Н, CF3СН2), 4,1 (м, 1H, CHBr), 4,2 (м, 2Н, СН2О); ЯМР 19F δ: -65 (т, 3F, 3JHF = 8 Гц, CF3).
1-Бром-3,3,3-трифторпропилацетат (5d).
Выход 70%. Т.кип. 30°С/10 Торр.
ЯМР 1H δ: 2,3 (с, 3H, CH3), 3,2-3,5 (уш.м, 2H, CH2), 7 (м, 1H, CHBr); ЯМР 19F δ: -65 (т, 3F, 3JHF = 8 Гц, CF3). Масс-спектр (M/Z, отнесение): 235[M+H]+, 191[C3H3BrF3O]+, 175[M-СH3CO2]+, 165[M-CF3]+, 155[C3H2BrF2]+, 129[CBrF2]+, 111[CHBrF]+, 95[C3H2F3]+, 91[C4HF2O]+, 75[C3HF2]+ , 64[C2H2F2]+, 51[CHF2]+, 43[C2H3O]+(100%), 36[C3]+.
4,4,4-Трифторбут-1-ен (6).
К суспензии 6,28 г (0,096 моль) порошка цинка в 100 мл воды присыпают при перемешивании и температуре 20°С 0,5 г (2,5 ммоль) Cu2Cl2 и смесь перемешивают в течение 10-15 минут. Далее при перемешивании и температуре 5°С добавляют по каплям 20 г (0,08 моль) 2-бром-4,4,4-трифторбутилацетата (5b) в 10 мл ацетона, реакционную смесь перемешивают в течение 1 часа при 20°С, нагревают до температуры 50÷80°С, отгоняя продукт реакции в охлаждаемый (0°С) приемник.
Полученный продукт очищают ректификацией.
Получают 7 г (выход 80%) 6. Т.кип. 10÷12°С.
ЯМР 1H δ: 3,7 (м, 2H, CF3CH2), 6,1 (м, 2H, CH=CH2), 6,6 (м, 1H, CH=CH2); ЯМР 19F δ: -68 (т, 3F, 3JHF = 14 Гц, CF3).
2-(2,2,2-Трифторэтил)оксиран (7).
Смесь 20 г (0,08 моль) 2-бром-4,4,4-трифторбутилацетата (5b), 7,7 г (0,24 моль) метанола и 0,1 мл 50% серной кислоты нагревают до кипения, медленно отгоняя на колонке Вигре метилацетат и метанол. Затем убирают колонку Вигре и отгоняют из реакционной смеси остатки метанола. Далее к реакционной смеси при перемешивании и температуре 100°С добавляют по каплям раствор 3,85 г (0,096 моль) гидроксида натрия в 5,8 мл воды отгоняя образующийся продукт.
Полученный продукт очищают ректификацией.
Получают 8 г (выход 80%) 7. Т.кип. 72÷75°С.
ЯМР 1H δ: 2,5-2,9 (уш.м, 2H, CF3CH2), 2,6-2,9 (уш.м, 2H, CF3CH2), 2,9, 3,2, 3,5 (м, 1H+1H+1H, оксирановый цикл); ЯМР 19F δ: -66 (т, 3F, 3JHF = 11 Гц, CF3).
(2E)-4,4,4-Трифторбут-2-ен-1-илацетат (9).
Смесь 58 г (0,23 моль) 2-бром-4,4,4-трифторбутилацетата (5b) и 33,6 г (0,46 моль) диэтиламина в 200 мл ацетона нагревают при перемешивании до слабого кипения в течение 4-5 часов. Смесь охлаждают, разбавляют трехкратным объемом воды, продукт экстрагируют 100 мл CH2Cl2, полученный раствор промывают холодной водой, растворитель отгоняют и остаток ректифицируют.
Получают 31 г (выход 80%) 9. Т.кип. 135÷138°С. Соотношение цис- и транс-изомеров составляет 1:9.
ЯМР 1H δ: 2,5 (c, 3H, CH3), 5,2 (уш.c, 2H, CH2), 6,4 (м, 1H, CH=), 7 (м, 1H, CH=); ЯМР 19F δ: -65 (уш.c, 3F, CF3). Масс-спектр (M/Z, отнесение): 169[M+H]+, 148[M-HF]+, 128[M-2HF]+, 109[M-CO2Me]+, 106[M-Ac-F]+, 95[C3H2F3]+, 89[M-AcO-HF]+, 77[C3H3F2]+, 69[CF3]+, 59[AcO]+, 51[CF2H]+, 43[C2F]+(100%), 39[C3H3]+.
2,2,2-Трифтор-N-(4,4,4-трифторбутил)ацетамид (10a).
В 6-ти литровую колбу, снабженную механической мешалкой, термометром, капельной воронкой и обратным холодильником наливают 2 л воды и 0,3 л ацетона, при перемешивании присыпают 260 г (3,97 моль) порошка цинка и 19 г (0,19 моль) хлористой меди. Смесь перемешивают в течение 15-20 минут, охлаждают до температуры 5°С и добавляют раствор 1000 г (3,31 моль) N-(2-бром-4,4,4-трифторбутил)трифторацетамида (5a) в 0,4 л ацетона с такой скоростью, чтобы температура смеси не поднималась выше 10°С. Внешнее охлаждение убирают, реакционную смесь перемешивают в течение 5-6 часов, при этом температура смеси поднимается до 30°С, и оставляют на ночь.
Затем реакционную смесь охлаждают до 15°С и при перемешивании медленно добавляют 0,6 л концентрированной соляной кислоты, не допуская бурного выделения водорода. Температуру смеси доводят до 20°С, перемешивание прекращают, дают реакционной смеси расслоиться, нижний слой отделяют, фильтруют через бумажный фильтр и промывают полуторакратным объемом 5% соляной кислоты. Получают продукт с примесью ацетона и 4,4,4-трифторбут-1-ена (6), которые удаляют отгонкой, нагревая смесь до 80÷100°С.
Получают 656 г (выход 80%) 10a, чистотой по ГЖХ 90%, который используют далее без дополнительной очистки.
Продукт можно дополнительно очистить перегонкой в вакууме.
Т.кип. 110÷115°С/10 Торр, кристаллический б/ц, Т.пл. 36°C.
ЯМР 1H δ: 2,3 (м, 2H, CF3CH2CH2), 2,6 (м, 2H, CF3CH2), 3,8 (м, 2H, CH2NH), 9,1 (уш.c, 1H, NH); ЯМР 19F δ: -78 (c, 3F, C(O)CF3), -68 (т, 3F, 3JHF = 11 Гц, CF3CH2).
4,4,4-Трифторбутиламин (11).
Смесь 656 г (2,65 моль) амида 10a (чистотой 90%, полученного по методу, описанному выше) и 160 г (3,2 моль) гидразингидрата, нагревают с отгонкой продукта кипящего 80÷105°С. Полученный дистиллят промывают равным объемом 30% водного раствора гидроксида натрия и перегоняют над твердым КОН.
Получают 268 г (выход 80%) 11, чистотой по ГЖХ 98%. Т.кип. 86÷87°С.
ЯМР 1H δ: 1,5 (м, 2H, CF3CH2CH2), 2,1 (м, 2H, CF3CH2), 2,6 (т, 2H, CH2NH2), 3,3 (уш.с, 2H, NH2); ЯМР 19F δ: -68 (т, 3F, 3JFH = 8 Гц, CF3).
5,5,5-Трифторамилацетат (10c).
К суспензии 59,6 г (0,91 моль) порошка цинка в 400 мл воды при перемешивании присыпают 3,96 г (0,04 моль) хлористой меди. Смесь перемешивают в течение 15-20 минут, охлаждают до температуры 5°С и добавляют раствор 200 г (0,76 моль) 3-бром-5,5,5-трифторпентилацетата (5c) в 140 мл ацетона с такой скоростью, чтобы температура смеси не поднималась выше 10°С. Внешнее охлаждение убирают, реакционную смесь перемешивают в течение 4-5 часов и оставляют на ночь.
Затем реакционную смесь охлаждают до 15°С и при перемешивании медленно добавляют 130 мл концентрированной соляной кислоты. Температуру смеси доводят до 20°С, перемешивание прекращают, дают реакционной смеси расслоиться, нижний слой отделяют, фильтруют через бумажный фильтр и перегоняют в вакууме.
Получают 112 г (выход 80%) 10c, который используют далее без дополнительной очистки.
ЯМР 1H δ: 2,0 (м, 4Н, СН2СН2CH2O), 2,3 (с, 3H, CH3), 2,5 (м, 2H, CF3CH2), 4,4 (т, 2Н, СН2О); ЯМР 19F δ: -68 (т, 3F, 3JHF = 11 Гц, CF3).
5,5,5-Трифторпентанол (12).
Смесь 100 г (0,54 моль) 5,5,5-трифторпентилацетата (10c), 52 г (1,62 моль) метанола и 0,5 мл 50% серной кислоты нагревают до кипения, медленно отгоняя на колонке Вигре метилацетат и метанол, остаток перегоняют.
Получают 69 г (выход 90%) 12. Т.кип. 150°С.
ЯМР 1H δ: 1,8 (м, 4H, CH2CH2CH2O), 2,3 (м, 2H, CF3CH2), 3,8 (т, 2H, CH2OH), 5,3 (c, 1H, CH2OH); ЯМР 19F δ: -68 (т, 3F, 3JHF = 8 Гц, CF3).
1,1,1-Трифтор-5-йодпентан (13).
В 4-x литровую колбу, снабженную механической мешалкой, термометром, капельной воронкой и обратным холодильником наливают 600 г (5,2 моль) 85% ортофосфорной кислоты, затем при перемешивании порциями присыпают 400 г (1,41 моль) фосфорного ангидрида, так чтобы температура смеси не поднималась выше 100°С. Смесь перемешивают в течение 40 минут, охлаждают до комнатной температуры и добавляют 459 г (2,77 моль) йодистого калия и 200 г (1,41 моль) 5,5,5-трифторпентанола (12). Далее реакционную смесь нагревают при перемешивании до температуры 105÷110°С в течении 5-6 часов и оставляют на ночь.
К реакционной смеси добавляют 1 л холодной воды, и 0,2 л хлористого метилена. Смесь перемешивают в течение 10 минут, затем отделяют нижний слой, промывают равным объемом холодной воды, раствором Na2SO3 до обесцвечивания. Растворитель отгоняют, и остаток перегоняют в вакууме 10 Торр, собирая фракцию, кипящую при 55÷60°С. Полученный продукт дополнительно очищают ректификацией.
Получают 302 г (выход 85%) 13, чистотой 98% по ГЖХ.
ЯМР 1H δ: 1,4, 1,6 (м, 2H+2H, CH2CH2CH2I), 1,8 (м, 2H, CF3CH2), 2,9 (т, 2H, CH2I); ЯМР 19F δ: -68 (т, 3F, 3JHF = 10 Гц, CF3).
4-(Ацетокси)-2-бром-3-(трифторметил)бутилацетат (15).
К раствору 100 г (0,58 моль) (2E)-1,4-диацетокси-2-бутена (14) в 100 мл ацетонитрила добавляют 0,63 г (0,01 моль) порошка меди. Раствор продувают аргоном и затем добавляют при перемешивании 123,5 г (0,58 моль) трифторметансульфонилбромида (1). Реакционную смесь перемешивают в течение двух дней при комнатной температуре, растворитель отгоняют и остаток перегоняют в вакууме 0,5 Торр. Продукт дополнительно очищают ректификацией.
Получают 130 г (выход 70%) аддукта 15 (смесь стереоизомеров). Т.кип. 95÷105°С/0,5 Торр.
ЯМР 1H δ: 2,2 (два с, 6H, С(O)CH3), 3,4-3,5 (м, 2H, CHBr+CF3CH), 4,6 (д, 4H, OCH2); ЯМР 19F δ: -68 (д, 3JHF = 8 Гц, CF3), -65 (д, 3JHF = 8 Гц, CF3) соотношением 1:1.
2-(Трифторметил)бут-3-енилацетат (16).
Получают аналогично 10c.
Выход 80%. Т.кип. 45÷55°С/10 Торр.
ЯМР 1H δ: 2,5 (c, 3H, C(O)CH3), 3,7 (м, 1H, CF3CH), 4,7 (м, 2H, OCH2), 5,9 (м, 2H, CH=CH2), 6,2 (м, 1H, CH=CH2); ЯМР 19F δ: -70 (д, 3F, 3JHF = 8 Гц, CF3).
2-(Трифторметил)бута-1,3-диен (17).
2-(Трифторметил)бутен-3-илацетат (16) пропускают по каплям в токе аргона через пирексную трубку, снабженную стеклянной насадкой, при 540÷550°С. Продукт реакции собирают в охлаждаемый (-10÷-20°С) приемник, соединенный с выходом реакционной трубы, после перегонки получают 17 чистотой около 90%.
Выход 35%. Т.кип. 35÷45°С.
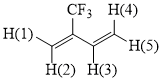
ЯМР 1H δ: 5,9 (д, 1H, H(5)), 6,2 (с, 1H, H(1)), 6,25 (д, 1H, H(4)), 6,35 (с, 1H, H(2)), 6,95 (дд, 1H, H(3)); ЯМР 19F δ: -68 (c, 3F, CF3). Масс-спектр (M/Z, отнесение): 122[M]+(100%), 121[M-H]+, 103[M-F]+, 102[M-HF]+, 101[M-HF-H]+, 83[CH2=C(CF)CH=CH]+, 77[C3H3F2]+, 75[CF2CCH]+, 69[CF3]+, 57[C3H2F]+, 53[M-CF3]+, 51[CF2H]+.
Метил-2-бром-4,4,4-трифторбутаноат (19a).
К раствору 20 г (0,23 моль) свежеперегнанного метилакрилата (18a) в 30 мл ацетонитрила добавляют 0,15 г (2,4 ммоль) порошка меди. Раствор продувают аргоном и затем добавляют при перемешивании 49 г (0,23 моль) трифторметансульфонилбромида (1). Реакционную смесь перемешивают в течение двух дней при комнатной температуре, растворитель отгоняют в вакууме 10 Торр в охлаждаемую (-50°С) ловушку и остаток перегоняют в вакууме 10 Торр. Продукт дополнительно очищают перегонкой в вакууме.
Получают 32 г (выход 60%) 19a. Т.кип. 50÷55°С/10 Торр.
ЯМР 1H δ: 2,85 (м, 1H, CF3CHA), 3,2 (м, 1H, CF3CHB), 3,8 (с, 3H, OCH3), 4,5 (т, 1H, CHBr); ЯМР 19F δ: -66 (т, 3F, 3JHF = 10 Гц, CF3). Масс-спектр (M/Z, отнесение): 235[M+H]+, 203[M-OMe]+, 175[M-CO2Me]+, 174[M-CO2Me-H]+, 155[M-CO2Me-HF]+, 135[M-Br-HF]+, 111[CHBrF]+, 95[M-CO2Me-Br-H]+, 91[CF2=CH-CH-CH3]+(100%), 77[M-CO2Me-F-Br]+, 69[CF3]+, 59[CO2Me]+, 51[CF2H]+.
2-Бром-4,4,4-трифторбутиронитрил (19b).
Получают аналогично 19a.
Выход 40%. Т.кип. 53÷57°С/10 Торр.
ЯМР 1H δ: 2,85-3,15 (м, 2H, CF3CH2), 4,65 (т, 1H, CHBr); ЯМР 19F δ: -66 (т, 3F, 3JHF = 9 Гц, CF3). Масс-спектр (M/Z, отнесение): 201[M]+, 182[M-F]+, 163[M-F-HF]+, 155[M-F-CN-H]+, 146[M-3F-H]+, 132[M-CF3]+, 129[M-CF3-3H]+, 122[M-Br]+(100%), 106[M-CF3-CN]+, 102[M-Br-HF]+, 94[M-Br-CN-2H]+, 75[M-Br-CN-H2-F]+, 69[CF3]+, 52[C2H2CN]+, 51[CF2H]+.
2-Бром-3-(трифторметансульфонил)пропаналь (21).
К раствору 15 г (0,268 моль) свежеперегнанного акролеина (20) в 20 мл ацетонитрила, содержащего 0,1 г (1,6 ммоль) порошка меди при перемешивании и температуре 25÷35°С прибавляют по каплям 57 г (0,268 моль) трифторметансульфонилбромида (1). Смесь перемешивают в течение 3 часов, летучие компоненты смеси отгоняются в вакууме 10 Торр в охлаждаемую (-50°С) ловушку и остаток перегоняют в вакууме 1 Торр, собирая фракцию кипящую 80÷95°С.
Получаю 57 г (выход 79%) 21, чистотой 90%. Т.кип. 80÷85°С/1 Торр.
ЯМР 1H δ: 3,85, 4,25 (ABкв, 2H, 2JHH = 30 Гц, CH2), 4,9 (т, 1H, CHBr), 9,4 (с, 1H, CHO); ЯМР 19F δ: -80 (с, 3F, CF3SO2). Масс-спектр (M/Z, отнесение): 269[M+H]+, 203[C4H3BrF3O]+, 189[M-Br]+, 135[C4H4BrO]+, 133[CF3SO2]+, 119[C4HF2S]+, 109[CHBrO]+, 108[CBrO]+, 107[C2H3Br]+, 106[C2H2Br]+(100%), 82[CF2S]+, 75[C3H4FO]+, 69[CF3]+, 65[C2H3F2]+, 55[C3H3O]+, 48[SO]+, 39[C3H3]+.
Реакция 1 со стиролом 22.
Получение (1-бром-3,3,3-трифторпропил)бензола (23) и 1-бром-2-(трифторметансульфонил)этилбензола (24).
A: Реакция на солнечном свету.
Готовят раствор 2 г (9,4 ммоль) трифторметансульфонилбромида (1) и 1 г (9,6 ммоль) свежеперегнанного стирола (22) в 6 мл гексана. Примерно 0,3 мл этого раствора запаивают в стеклянную ампулу из молибденового стекла (диаметром 3 мм и длиной 100 мм) и выдерживают на солнечном свету в течение 1 дня на подоконнике. Затем ампулу помещают в стандартную 5 мм ЯМР ампулу, добавляют в качестве внешнего стандарта CDCl3 и реакционную смесь анализируют методом ЯМР 19F.
По данным ЯМР 19F смесь содержит 34% 23 (δ: -65, т, 3JHF = 9 Гц,) и 65% 24 (δ: -80, с).
B: Реакция в присутствии меди в растворе ацетонитрила.
Реакцию проводят по общей методике при температуре 25÷30°С. Реакционную смесь анализируют методом ЯМР 19F.
По данным ЯМР 19F смесь содержит 25% 23 и 75% 24.
Реакционную смесь перегнали в вакууме масляного насоса, собирая летучие продукты в охлаждаемую ловушку (-78°С). При перегонке вакуум колебался в пределах 0,5-10 Торр из-за дегазации перегоняемой смеси. Полученный дистиллят содержит смесь продуктов 23, 24 и 25 (последний, очевидно, образуется в результате дегидробромирования соединения 24). Смесь повторно перегоняют, собирая фракцию, выкипающую в интервале 110÷151°С/10 Торр. По данным ЯМР 1H и 19F смесь содержит 23% 23, 34% 24 и 43% 25, а также незначительное количество дибромида стирола.
При стоянии данной смеси выкристаллизовывается винилсульфон 25.
(E)-PhCH=CHSO2CF3 (25).
ЯМР 1H δ: 6,85 (д, 1H, PhCH=), 7,93 (д, 1H, =CHSO2), 7,45-7,65 (м, 5H, Ph); ЯМР 19F δ: -79 (c, CF3). Масс-спектр (M/Z, отнесение): 236[M]+, 196[C6H5CCSO2CF]+, 167[M-CF3]+, 151[C6H5CHCHSO]+, 134[C6H5CHCHCF]+, 119[C6H5CHCHO]+, 103[M-SO2CF3]+(100%), 93[C6H5O]+, 89[C6H5C]+, 77[C6H5]+, 69[CF3]+, 51[CF2H]+.
Список литературы
- F. Ooms, R. Frederick, F. Durant, J.P. Petzer, N. Castagnoli Jr., C. J Van der Schyf, J. Wouters, Bioorg.Med.Chem.Lett., 2003, 13, 69-73.
- J. Reniers, S. Robert, R. Frederick, B. Masereel, S. Vincent, J. Wouters, Bioorg.Med.Chem., 2011, 19, 134-144.
- U. Gerlach, J. Brendel, H.-J. Lang, E.F. Paulus, K. Weidmann, A. Bruggemann, A.E. Busch, H. Suessbrich, M. Bleich, R. Greger, J.Med.Chem., 2001, 44, 3831-3837.
- R.W. Winter, J.X. Kelly, M.J. Smilkstein, R. Dodean, D. Hinrichs, M.K. Riscoe, Exp Parasitol., 2008, 118, 487-497.
- T. Rodrigues, F. Lopes, R. Moreira, Curr.Med.Chem., 2010, 17, 929-956.
- F.A. Romero, S.M. Vodonick, K.R. Criscione, M.J. McLeish, G.L. Grunewald, J.Med.Chem., 2004, 47, 4483-4493.
- M.K. Riscoe, R.W. Winter, J.X. Kelly, M.J. Smilkstein, D.J. Hinrichs, US Pat. № 7,829,578 B1 (2010).
- Q. Yang, B. Lorsbach, G. Whiteker, G. Roth, C. DeAmicis, D.I. Knueppel, A.M. Buysse, K. Gray, X. Li, J.M. Muhuhi, R. Ross Jr., D.E. Podhorez, Y. Zhang, US Pat. № 9,414,594 B2 (2016).
- W. Hierse, N. Ignatyev, M. Seidel, E. Montenegro, P. Kirsch, A. Bathe, US Pat. № 8,067,625 B2 (2011).
- N. Terasawa, H. Monobe, K. Kiyohara, J.Fluor.Chem., 2006, 127, 954-961.
- A.R. Vanover, J.D. Necessary, K.B. Chandalia, J.W. Reisch, J.M. O’Connor, K. Delaney, P.R. Miller, US Pat. № 5,002,678 (1991).
- H. Buchwald, A. Brackmann, B. Raszkowski, US Pat. № 5,304,321 (1994).
- B. Bidlingmeyer, Q. Wang, US Pat. № 7,125,492 B2 (2006).
- K.J. Cross, US Pat. № 8,939,208 B2 (2015).
- M. Antinolo, E. Jimenez, J. Albaladejo, Environ.Sci.Technol., 2011, 45, 4323-4330.
- W.E. Hanford, R.M. Joyce Jr., US Pat. № 2,440,800 (1948).
- M. Van Der Puy, A. Thenappan, US Pat. № 5,777,184 (1998).
- J.-A. Ma, D. Cahard, JFC, 2007, 128, 975-996.
- A. Studer, Angew.Chem.I.E., 2012, 51, 8950-8958.
- S. Barata-Vallejo, Al Postigo, Coord.Chem.Rev., 2013, 257, 3051-3069.
- H. Egami, M. Sodeoka, Chem.Eur.J., 2014, 53, 8294-8308.
- S. Barata-Vallejo, B. Lantano, Al Postigo, Chem.Eur.J., 2014, 20, 16806-16829.
- W. Zeng, F. Chen, Chin.J.Appl.Chem., 2014, 31, 627-641.
- C. Ni, M. Hu, J. Hu, Chem.Rev., 2015, 115, 765-825.
- X.-H. Xu, K. Matsuzaki, N. Shibata, Chem.Rev., 2015, 115, 731-764.
- P. Gao, X.-R. Song, X.-Y. Liu, Y.-M. Liang, Chem.Eur.J., 2015, 21, 7648-7661.
- R.N. Haszeldine, JCS, 1949, 2856-2861.
- N.O. Brace, JOC, 1963, 28, 3093-3102.
- J.D. Park, F.E. Rogers, J.R. Lacher, JOC, 1961, 26, 2089-2095.
- J. Ignatowska, W. Dmowski, JFC, 2007, 128, 997-1006.
- T. Kitazume, N. Ishikawa, JACS, 1985, 107, 5186-5191.
- S. Mukhopadhyay, H.K. Nair, H.S. Tung, M. Van Der Puy, US Pat. № 0245773 A1 (2005).
- V.A. Petrov, C.G. Krespan, JFC, 2000, 102, 199-204.
- G.V.D. Tiers, US Pat. № 2,965,659 (1960).
- N. Kamigata, T. Fukushima, M. Yoshida, J.Chem.Soc.Chem.Commun., 1989, 1559-1560.
- N. Kamigata, M. Yoshida, H. Sawada, M. Nakayama, US Pat. № 5,118,879 (1992).
- H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang, S. Yu, Eur.JOC, 2012, 18, 15158-15166.
- H. Jiang, Y. Cheng, Y. Zhang, S. Yu, Eur.JOC, 2013, 5485-5492.
- S.H. Oh, Y.R. Malpani, N. Ha, Y.-S. Jung, S.B. Han, Org.Lett., 2014, 16, 1310-1313.
- X.-J. Tang, W.R. Dolbier Jr., Angew.Chem.I.E., 2015, 54, 4246-4249.
- D.B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B.M. Bhanage, O. Reiser, Angew.Chem.I.E., 2015, 54, 6999-7002.
- C. Zhang, Adv.Synth.Catal., 2014, 356, 2895-2906.
- Y. Lu, Y. Li, R. Zhang, K. Jin, C. Duan, JFC, 2014, 161, 128-133.
- B. Yang, X.-H. Xu, F.-L. Qing, Org.Lett., 2015, 17, 1906-1909.
- A. Deb, S. Manna, A. Modak, T. Patra, S. Maity, D. Maiti, Angew.Chem.I.E., 2013, 125, 9929-9932.
- D.J. Wilger, N.J. Gesmundo, D.A. Nicewicz, Chem.Sci., 2013, 4, 3160-3165.
- Q. Lefebvre, N. Hoffmann, M. Rueping, Chem.Commun., 2016, 52, 2493-2496.
- W.-Y. Huang, L. Lu, Chin.J.Chem., 1992, 10, 268-273.
- CN Pat. № 1730127 B (2010).
- W.-Y. Huang, J.-L. Chen, ActaChim.Sin.Eng.Ed., 1988, 150-154.
- Y.-F. Zhang, L. Lu, W.-Y. Huang, ActaChim.Sin.Eng.Ed., 1989, 376-384.
- X.-K. Jiang, G.-Z. Ji, J.R.-Y. Xie, JFC, 1996, 79, 133-138.
- L. Chu, F.-L. Qing, Org.Lett., 2012, 14, 2106-2109.
- X. Wu, L. Chu, F.-L. Qing, Angew.Chem.I.E., 2013, 52, 2198-2202.
- F. Wang, X. Qi, Z. Liang, P. Chen, G. Liu, Angew.Chem.I.E., 2014, 53, 1881-1886.
- M. Fu, L. Chen, Y. Jiang, Z.-X. Jiang, Z. Yang, Org.Lett., 2016, 18, 348-351.
- J. Charpentier, N. Fruh, A. Togni, Chem.Rev., 2015, 115, 650-682.
- M. Van Der Puy, T.R. Demmin, G.V.B. Madhavan, A. Thenappan, H.S. Tung, JFC, 1996, 76, 49-54.
- H.K. Nair, A.J. Poss, US Pat. № 6,476,279 B2 (2002).
- Z.-Y. Long, Q.-Y. Chen, Tetrahed.Lett., 1998, 39, 8487-8490.
- M. Huang, L. Li, Z.-G. Zhao, Q.-Y. Chen, Y. Guo, Synthesis, 2015, 47, 3891-3900.
- X.-J. Tang, C.S. Thomoson, W.R. Dolbier Jr., Org.Lett., 2014, 16, 4594-4597.
- G.K.S. Prakash, A.K. Yudin, Chem.Rev., 1997, 97, 757-786.
- R.O. Bolt, R.M. Joyce, Chem.Eng.News, 1947, 25, 1866-1867.
- F. Minisci, Acc.Chem.Res., 1975, 8, 165-171.
- M. Asscher, D. Vofsi, JCS, 1964, 4962-4971.
- L.I. Zakharkin, G.G. Zhigareva, Zh.Org.Khim., 1973, 9, 891-895.
- Y. Amiel, JOC, 1974, 39, 3867-3870.
- N. Roques, US Pat. № 2004/0147789 A1 (2004).
- J.M. Tedder, J.C. Walton, Tetrahedron, 1980, 36, 701-707.
- B. Giese, Angew.Chem.I.E., 1983, 22, 753-764.
Статья рекомендована к публикации членом редколлегии к.х.н.А. А. Тютюновым
Fluorine Notes, 2016, 109, 1-2
