Received: March, 2016
DOI 10.17677/fn20714807.2016.02.02
Fluorine Notes, 2016, 105, 3-4
Lewis Acid-Catalyzed Selective Mono-fluorination of Malonates Using Me-NFSI
Kazunobu Fukushi, a Etsuko Tokunaga,b Yuji Sumii,a Takumi Kagawa,c Noritaka Nagasaki,c and Norio Shibataa,b*
aDepartment of Nanopharmaceutical Sciences, and bDepartment of Frontier
MaterialsaDepartment of Nanopharmaceutical Sciences, and bDepartment
of Frontier Materials,
Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555,
Japan.
E-mail: nozshiba@nitech.ac.jp; Fax:
+81-52-735-7543
cTOSOH F-TECH, Inc., 4988, Kaisei-cho, Shunan, Yamaguchi 746-0006, Japan
Abstract: The Lewis acid-catalyzed selective mono-fluorination of malonates by Me-NFSI is disclosed. The sterically demanding Lewis acid of Ti(OtBu)4 is a key for this catalytic transformation and desired mono-fluorinated malonates were obtained in moderate to good yields with a small amount of di-fluorinates. The substrate-scope is also discussed.
Keywords: Electrophilic fluorination, malonate, Lewis acid, shelf-stable reagent
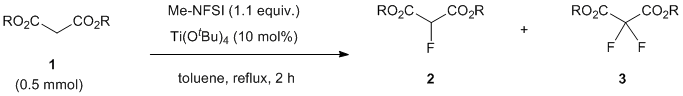
The selective synthesis of mono-fluorinated compounds [1] has gained considerable attention over the last decades in the fields of medicinal chemistry and material sciences, since the introduction of one fluorine atom into original compounds often dramatically improved or changed the properties of molecules without much difference to their basic chemical structures [2]. In this endeavor, the direct electrophilic fluorination reaction of nucleophiles has been extensively explored from the viewpoint of both catalysts and reagents [3]. Gaseous fluorine (F2) is a classic, very fundamental reagent for the electrophilic fluorination reaction but it is highly toxic and too reactive, making its reactivity difficult to control [4]. Perchloryl fluoride (FClO3) [5] and trifluoromethyl hypofluorite (CF3OF) [6] are also reactive reagents for electrophilic fluorination, but they are highly likely to be explosive. In this context, there has been a growing demand for developing shelf-stable reagents for direct electrophilic fluorination with a view to establishing selective fluorination under mild conditions [7]. Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor®) [8] and N-fluorobenzenesulfonimide (NFSI) [9] have been developed for this purpose and became two of the most popular reagents in this field (Figure 1). Although these two reagents are easily handled with ideal wide reactivity, it is not convenient for large-scale use due to their poor atom-economic transformation. Recently, we reported that N-fluoromethanesulfonimide (F-N(SO2Me)2, Me-NFSI) is an atom-economic alternative to NFSI, showing higher fluorination reactivity of β-keto esters, oxindoles and substituted malonates under Lewis acid-catalysis and non-catalysis [10] (Figure 1c). In particular, Me-NFSI has advantages over NFSI for the electrophilic fluorination of active methine compounds under acid-catalysis. In order to expand the utility of Me-NFSI, we were next interested in the preparation of mono-fluorinated malonates.
Figure 1. Shelf-stable reagents for electrophilic fluorination
Monofluoromalonates are versatile building blocks for the synthesis of pharmaceuticals and agrochemicals [11] such as difluprednate (Durezol®), Fluoxastrobin (Pestanal®) and TAK-733. Monofluoromalonates are commonly accessed by direct fluorination of malonates using electrophilic-type reagents such as AcOF [12], xenon difluoride [13], and shelf-stable fluorinating reagents of 1-fluoro-2-pyridone [14] and N-fluoropyridinium salts [15]. However, the fluorination of malonates always results in a mixture of mono- and di-fluorination products. Therefore, the selective mono-fluorination of malonates that can suppress over-fluorination is of great importance, although stepwise methods for the preparation of mono-fluoro malonates such as electrophilic fluorination of silyl ketene acetals [16] and 2-formyl malonates [17] are alternative means to achieve this. In 1998, Chambers and co-workers reported selective mono-fluorination by F2 under Lewis acid conditions [18]. In this method, Lewis acids coordinate malonates and enable the control of mono-fluorination. In 2014, Hu and co-workers reported mono-fluorination by an electrophilic fluorinating reagent, Selectfluor® in the presence of a stoichiometric amount of Lewis acid [19]. Recently, Sandford and Harsanyi improved the original Chambers report for the mono-fluorination reaction with F2 using Lewis acid to provide mono-fluoromalonates in up to 98% yield [20]. The fact that the direct electrophilic fluorination of malonates suffers fundamentally from the lack of mono- versus di-selectivity due to the higher reactivity of mono-fluorinated malonates toward electrophilic fluorination has led the nucleophilic approach to have market success in the treatment of 2-chloromaronate with hydrogen fluoride (HF) [21, 22, 23]. Kitamura and co-workers provided a unique solution for the nucleophilic mono-selective fluorination of malonates using hypervalent iodine reagent and HF [24]. Although all these methods are useful due to their high mono-selectivity during the fluorination of malonates, it is still attractive to develop novel approaches for this purpose. As mentioned earlier in the introduction, we disclosed that Me-NFSI is an atom-economic fluorination reagent [10, 25]. To extend the utility of Me-NFSI, we executed the mono-fluorination of malonates using Me-NFSI under Lewis acid-catalysis.
Using previous conditions [10], we first examined the mono-fluorination of diethyl malonate (1a) using Me-NFSI in the presence of a catalytic amount of Ti(OiPr)4 under reflux. The 19F-NMR study of the crude material after the reaction indicated that the fluorination reaction was more complicated than expected. The chemical shift of mono-fluorinated product 2a and di-fluorinated product 3a in the crude 19F-NMR spectra was observed at –195.55 ppm (doublet) and at –112.73 ppm (singlet), respectively. However, we noticed that other signals having similar patterns to the mono- and di-fluorinated products 2a and 3a were also observed at around –195 ppm and –110 ppm (Figure 2). Further attempts to isolate and purify the two by-products from the mixture failed.
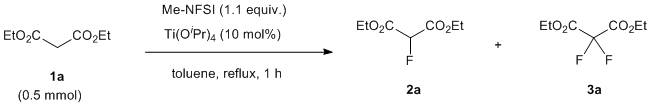
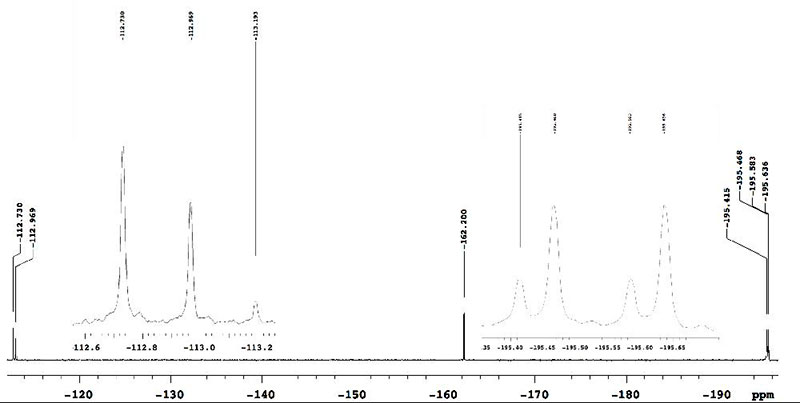
Figure 2. Crude 19F-NMR spectrum of Et ester using Ti(OiPr)4
We assumed that the byproducts could result in the transesterification of substrate with isopropyl alcohol generated from Ti(OiPr)4. We next examined the fluorination reaction using benzyl malonate and methyl malonate under the same conditions, and a similar small amount of byproducts was again observed in the crude 19F-NMR. On the other hand, in the case of isopropyl malonates (1e), the reaction proceeded very cleanly to provide mono-fluorinated product 2e (–195.42 ppm, doublet, J = 47.4 Hz) and di-fluorinated product 3e (–113.17 ppm, singlet) (Figure 3).
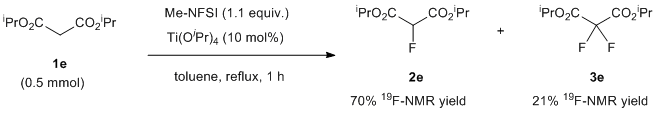
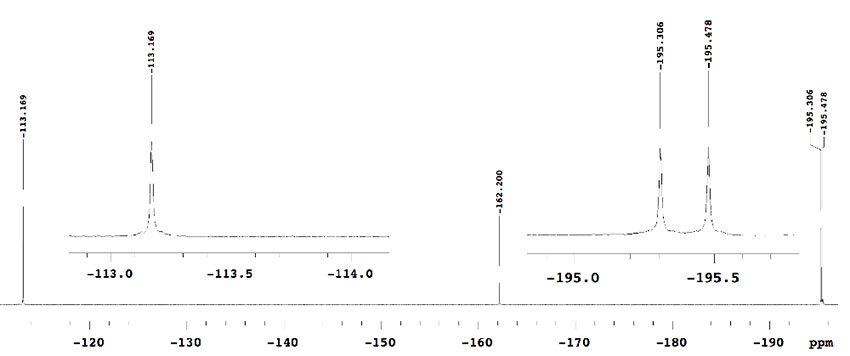
Figure 3. Crude 19F-NMR spectrum of iPr ester using Ti(OiPr)4
These results obviously indicate that the minor products in the fluorination of 1a-d should be fluorinated isopropyl esters of malonates formed by transesterification induced by Ti(OiPr)4. This problem was solved by the use of the sterically demanding Lewis acid of Ti(OtBu)4 instead of Ti(OiPr)4. Thus, we next examined the fluorination of 1a by Me-NFSI in the presence of Ti(OtBu)4. As expected, desired mono-fluorinated product 2a with di-fluorinated 3a were produced under the same conditions (Figure 4).
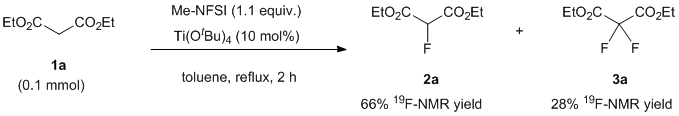
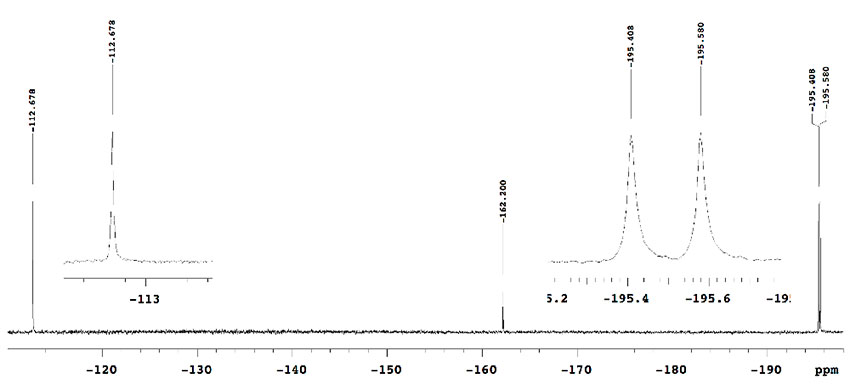
Figure 4. Crude 19F-NMR spectrum of Et ester using Ti(OtBu)4
With this condition in hand, we examined the mono-fluorination of malonates having a variety of ester groups (Table 1). Ethyl malonate (1a) was converted into mono-fluorinated product 2a in good yield (60% NMR yield, and 45% isolated yield) with a small amount of di-fluorinated product 3a (15% NMR yield), without the formation of any transesterification products in crude 19F-NMR (entry 1). On the other hand, the fluorination of methyl malonate 1b was not very slow under the same conditions and initial 1b was recovered (entry 2). Increasing the amount of Ti(OtBu)4 to 30-50 mol% increased the formation of fluorination products 2b and 3b, although a trace amount of potential transesterification products were detected by 19F-NMR at the expense of Ti(OtBu)4 (entries 3 and 4). Methyl mono-fluorinated malonate 2b was isolated in 30% yield under 50 mol% of Ti(OtBu)4 (entry 4). Butyl mono-fluorinated product 2c was isolated in 58% yield by the fluorination of malonic n-Bu diester 1c in the presence of 10 mol% of catalyst (entry 5). Benzyl malonate 1d and isopropyl malonate 1e were nicely fluorinated under the same conditions to afford mono-fluorinated products 2d and 2e in moderated yields (2d: 49% 2e: 48%) without the detection of any transesterification (entries 6 and 7). In the case of isopropyl diester, Ti(OiPr)4 was more effective than Ti(OtBu)4, affording mono-fluorinated 2e with a higher yield of 65% (entry 8). In all cases, the formation of small amounts of possible di-fluorinated products 3 was detected by 19F NMR spectroscopy of the corresponding reaction mixtures, but subsequent column chromatography did not allow for the separation of these compounds except for butyl ester 3c (12% isolated yield, entry 5).
Table 1. Fluorination of Ethyl Malonate (1a) by MeNFSI
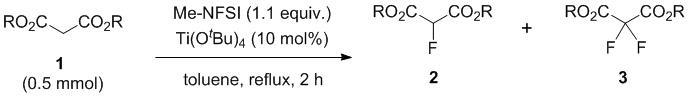
|
Entry |
R |
1 |
NMR yield (%)a |
Isolated yield (%) |
||
|
2 |
3 |
2 |
3 |
|||
|
1 |
Et |
1a |
60 |
15 |
45 |
0 |
|
2 |
Me |
1b |
26 |
2 |
- |
- |
|
3 b |
Me |
1b |
40 |
8 |
- |
- |
|
4c,d |
Me |
1b |
45 |
11 |
30 |
0 |
|
5 |
n-Bu |
1c |
65 |
13 |
58 |
12 |
|
6 |
Bn |
1d |
71 |
11 |
49 |
0 |
|
7 |
iPr |
1e |
51 |
14 |
48 |
0 |
|
8e |
iPr |
1e |
70 |
21 |
65 |
0 |
a) Yield was determined by 19F-NMR analysis of the crude mixture with CF3Ph as an internal standard. b) 30 mol% of Ti(OtBu)4 was used. c) 50 mol% of Ti(OtBu)4 was used. d) Transesterification product peaks were observed. e) Ti(OiPr)4 was used instead of Ti(OtBu)4, and the reaction time was 6 h.
In conclusion, we demonstrated the selective mono-fluorination of malonates 1 using an atom-economic fluorinating reagent, Me-NFSI, in the presence of Ti(OtBu)4. Transesterification was observed by Ti(OiPr)4-catalyzed fluorination but this side reaction was inhibited by the use of a steric Lewis acid of Ti(OtBu)4 instead [26]. Thus, the combination of Me-NFSI with a catalytic amount of Ti(OtBu)4 under reflux conditions was effective for transesterification-free selective mono-fluorination of malonates in moderated yields. The application of this method for the selective mono-fluorination of a variety of active methylene compounds is now under investigation.
Experimental
All reactions were performed in oven-dried glassware under a positive pressure of nitrogen. Solvents were transferred via syringe and were introduced into the reaction vessels though a rubber septum. Column chromatography was carried out on a column packed with silica-gel 60N spherical neutral size 63-210. The 1H-NMR (300 MHz), 19F-NMR (282 MHz), and 13C-NMR (75.5 MHz and 100.7 MHz) spectra for the CDCl3 solution were recorded on a Varian Mercury 300 and Bruker Avance 400. Chemical shifts (δ) are expressed in ppm downfield from internal TMS (δ = 0.00), CHCl3 (δ = 77.0) or C6F6 (δ = 162.2). Mass spectra were recorded on a SHIMADZU LCMS-2020 (ESI-MS). High resolution mass spectrometries were recorded on a Waters Synapt G2 HDMS (ESI-MS). Infrared spectra were recorded on a JASCO FT/IR-4100 spectrometer. Me-NFSI was synthesized according to Fukushi et al. [10]. Other reagents and solvents were purchased.
General procedure for the fluorination of malonates
To a stirred mixture of malonates 1 (0.5 mmol) and Me-NFSI (105.6 mg, 0.55 mmol, 1.1 equiv.) in toluene (5.0 mL, 0.1 M), Ti(OtBu)4 (20 μL, 0.050 mmol, 10 mol%) was added under a nitrogen atmosphere and stirred at reflux for 2 h. The reaction mixture was cooled to room temperature, quenched with a saturated NaHCO3 aqueous solution, and extracted three times with EtOAc. The organic layer was washed with brine, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/CH2Cl2) to give fluorinated malonates 2.
Diethyl 2-fluoromalonate (2a)
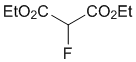
To a stirred mixture of diethyl malonate 1a (80.1 mg, 76 μL, 0.5 mmol) and Me-NFSI (105.6 mg, 0.55 mmol, 1.1 equiv.) in toluene (5.0 mL, 0.1 M), Ti(OtBu)4 (20 μL, 0.050 mmol, 10 mol%) was added under nitrogen atmosphere and stirred at reflux for 2 h. The reaction mixture was cooled to room temperature, quenched with a saturated NaHCO3 aqueous solution, and extracted three times with EtOAc. The organic layer was washed with brine, dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/CH2Cl2 = 1/1) to give fluorinated malonic diester 2a (40.3 mg, 45%) as a colorless oil.
1H-NMR (CDCl3, 300 MHz) δ 5.28 (d, J = 48.2 Hz, 1H), 4.34 (q, J = 7.1 Hz, 4H), 1.34 (t, J = 7.1 Hz, 6H); 19F-NMR (CDCl3, 282 MHz) δ 195.58 (d, J = 48.3 Hz, 1F); MS (ESI, m/z) 201 [M+Na]+.
This compound has been previously prepared and characterized [24].
Dimethyl 2-fluoromalonate (2b)
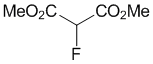
To a stirred mixture of dimethyl malonate 1b (66.1 mg, 57 μL, 0.5 mmol) and Me-NFSI (105.6 mg, 0.55 mmol, 1.1 equiv.) in toluene (5.0 mL, 0.1 M), Ti(OtBu)4 (20 μL, 0.050 mmol, 10 mol%) was added under nitrogen atmosphere and stirred at reflux for 2 h. The reaction mixture was cooled to room temperature, quenched with a saturated NaHCO3 aqueous solution, and extracted three times with EtOAc. The organic layer was washed with brine, dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/CH2Cl2 = 1/1) to give fluorinated malonic diester 2b (22.5 mg, 30%) as a colorless oil.
1H-NMR (CDCl3, 300 MHz) δ 5.34 (d, J = 48.0 Hz, 1H), 3.88 (s, 6H); 19F-NMR (CDCl3, 282 MHz) δ 195.72 (d, J = 48.5 Hz, 1F); MS (ESI, m/z) 189 [M+K]+.
This compound has been previously prepared and characterized [20].
Di-n-butyl 2-fluoromalonate (2c) and di-n-butyl 2,2-difluoromalonate (3c)
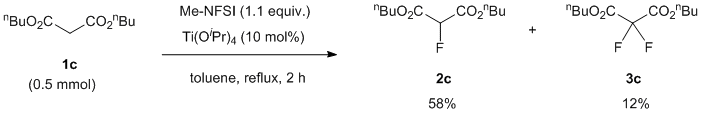
To a stirred mixture of di-n-butyl malonate 1c (108.1 mg, 110 μL, 0.5 mmol) and Me-NFSI (105.6 mg, 0.55 mmol, 1.1 equiv.) in toluene (5.0 mL, 0.1 M), Ti(OtBu)4 (20 μL, 0.050 mmol, 10 mol%) was added under nitrogen atmosphere and stirred at reflux for 2 h. The reaction mixture was cooled to room temperature, quenched with a saturated NaHCO3 aqueous solution, and extracted three times with EtOAc. The organic layer was washed with brine, dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/CH2Cl2 = 1/1) to give fluorinated malonic diester 2c (68.3 mg, 58%) and di-fluorinated malonic diester 3c (15.4 mg, 12%), both as colorless oils.
Di-n-butyl 2-fluoromalonate (2c)
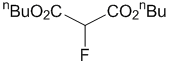
1H-NMR (CDCl3, 300 MHz) δ 5.26 (d, J = 48.3 Hz, 1H), 4.24 (t, J = 6.0 Hz, 4H), 1.69-1.60 (m, 4H), 1.40-1.33 (m, 4H), 0.91 (t, J = 7.2 Hz, 6H); 19F-NMR (CDCl3, 282 MHz) δ 195.40 (d, J = 47.4 Hz, 1F); MS (ESI, m/z) 257 [M+Na].
This compound has been previously prepared and characterized [20].
Di-n-butyl 2,2-difluoromalonate (3c)
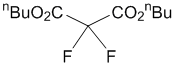
1H-NMR (CDCl3, 300 MHz) δ 4.33 (t, J = 6.3 Hz, 4H), 1.75-1.65 (m, 4H), 1.47-1.39 (m, 4H), 0.95 (t, J = 7.2 Hz, 6H); 19F-NMR (CDCl3, 282 MHz) δ 112.57 (s, 2F); 13C-NMR (CDCl3, 100.7 MHz) δ 160.85 (t, J = 30.9 Hz), 106.08 (t, J = 261.1 Hz), 67.52, 30.17, 18.78, 13.52; IR (neat) 2965, 2947, 2881, 1781, 1271, 1160, 1070 cm-1; MS (ESI, m/z) 275 [M+Na]+; HRMS (ESI) C11H18F2NaO4 [M+Na]+ 275.1071 calcd for 275.1070.
Dibenzyl 2-fluoromalonate (2d)
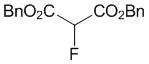
To a stirred mixture of dibenzyl malonate 1c (142.2 mg, 123 μL, 0.5 mmol) and Me-NFSI (105.6 mg, 0.55 mmol, 1.1 equiv.) in toluene (5.0 mL, 0.1 M), Ti(OtBu)4 (20 μL, 0.050 mmol, 10 mol%) was added under nitrogen atmosphere and stirred at reflux for 2 h. The reaction mixture was cooled to room temperature, quenched with a saturated NaHCO3 aqueous solution, and extracted three times with EtOAc. The organic layer was washed with brine, dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/CH2Cl2 = 1/1) to give fluorinated malonic diester 2d (74.3 mg, 49%) as a colorless oil.
1H-NMR (CDCl3, 300 MHz) δ 7.36-7.30 (m, 10H), 5.35 (d, J = 47.9 Hz, 1H), 5.24 (s, 4H); 19F-NMR (CDCl3, 282 MHz) δ 195.56 (d, J = 47.4 Hz, 1F); MS (ESI, m/z) 325 [M+Na]+.
This compound has been previously prepared and characterized [24].
Di-iso-propyl 2-fluoromalonate (2e)
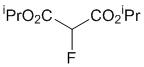
To a stirred mixture of di-iso-propyl malonate 1e (94.1 mg, 95 μL, 0.5 mmol) and Me-NFSI (105.6 mg, 0.55 mmol, 1.1 equiv.) in toluene (5.0 mL, 0.1 M), Ti(OtBu)4 (20 μL, 0.050 mmol, 10 mol%) or Ti(OiPr)4 (15 μL, 0.050 mmol, 10 mol%) were added under nitrogen atmosphere and stirred at reflux for 2 h or 6 h. The reaction mixture was cooled to room temperature, quenched with a saturated NaHCO3 aqueous solution, and extracted three times with EtOAc. The organic layer was washed with brine, dried with Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/CH2Cl2 = 1/1) to give fluorinated malonic diester 2e (49.3 mg, 48%; Ti(OtBu)4) or (67.0 mg, 65%; Ti(OiPr)4) as a colorless oil.
1H-NMR (CDCl3, 300 MHz) δ 5.20 (d, J = 48.3 Hz, 1H), 5.19 (q, J = 6.3 Hz, 2H), 1.31 (t, J = 6.3 Hz, 6H); 19F-NMR (CDCl3, 282 MHz) δ 195.42 (d, J = 47.4 Hz, 1F); 13C-NMR (CDCl3, 75.5 MHz) δ 163.55 (d, J = 24.6 Hz), 85.41 (d, J = 195.3 Hz), 70.74, 21.51 (d, J = 4.6 Hz); IR (neat) 2986, 2940, 1772, 1748, 1256, 1101, 988 cm-1; MS (ESI, m/z) 229 [M+Na]+; HRMS (ESI) C9H15FNaO4 [M+Na]+ 229.0852 calcd for 229.0857.
Acknowledgement
This research was financially supported in part by the Platform for Drug Discovery, Informatics, and Structural Life Science from MEXT Japan, the Advanced Catalytic Transformation (ACT-C) from the Japan Science and Technology (JST) Agency, the Kobayashi International Foundation and Hoansha Foundation.
References and Notes
- (a) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2013; (b) J.-A. Ma, D. Cahard, Chem. Rev. 2008, 108, PR1; (c) K. L. Kirk, Org. Process Res. Dev. 2008, 12, 305; (d) T. Liang, C. N. Neumann, T. Ritter, Angew. Chem., Int. Ed. 2013, 52, 8214; (e) J. Wu, Tetrahedron Lett. 2014, 55, 4289; (f) A. J. Cresswell, S. G. Davies, P. M. Roberts, J. E. Thomson, Chem. Rev. 2015, 115, 566.
- (a) B. E. Smart, J. Fluorine Chem. 2001, 109, 3; (b) P. Jeschke, ChemBioChem 2004, 5, 570; (c) K. Müller, C. Faeh, F. Diederich, Science 2007, 317, 1881; (d) S. Purser, P. R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 2008, 37, 320; (e) W. K. Hagmann, J. Med. Chem. 2008, 51, 4359; (f) R. Berger, G. Resnati, P. Metrangolo, E. Weber, J. Hulliger, Chem. Soc. Rev. 2011, 40, 3496; (g) M. Bremer, P. Kirsch, M. Klasen-Memmer, K. Tarumi, Angew. Chem., Int. Ed. 2013, 52, 8880; (h) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly, N. A, Meanwell, J. Med. Chem. 2015, 58, 8315.
- (a) S. Lectard, Y. Hamashima, M. Sodeoka, Adv. Synth. Catal. 2010, 352, 2708; (b) G. Valero, X. Companyó, R. Rios, Chem. Eur. J. 2011, 17, 2018; (c) T. Umemoto, J. Fluorine Chem. 2014, 167, 3; (d) M. G. Campbell, T. Ritter, Chem. Rev. 2015, 115, 612; (e) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme, J.-F. Paquin, Chem. Rev. 2015, 115, 9073.
- (a) Efficient Preparations of Fluorine Compounds, ed. H. W. Roesky, Wiley, New Jersey, 2012; (b) G. Sandford, J. Fluorine Chem. 2007, 128, 90.
- (a) A. Engelbrecht, H. Atzwanger, J. Inorg. Nucl. Chem. 1956, 2, 348;
(b) C. E. Inman, R. E. Oesterling, E. A. Tyczkowski, J. Am. Chem. Soc. 1958,
80, 6533; (c) H. Gershon, S. G. Schulman, A. D. Spevack, J. Med. Chem. 1967, 10, 536; (d) G. Kh. Khisamutdinov, L. V. Okhlobystina,
andA. A. Fainzil´berg Russ. Chem. Bull., Int. Ed. 2009, 58, 2182. - (a) K. B. Kellogg, G. H. Cady, J. Am. Chem. Soc. 1948, 70, 3986; (b) S. Rozen, Chem. Rev. 1996, 96, 1717; (b) W. Navarrini, F. Venturini, M. Sansotera, M. Ursini, P. Metrangolo, G. Resnati, M. Galimberti, E. Barchiesi, P. Dardani, J. Fluorine Chem. 2008, 129, 680; (c) L. Du, D. D. DesMarteau, V. Tortelli, M. Galimberti, J. Fluorine Chem. 2009, 130, 830; (d) F. Venturini, W. Navarrini, A. Famulari, M. Sansotera, P. Dardani, V. Tortelli, J. Fluorine Chem. 2012, 140, 43.
- General review of N–F type reagents for electrophilic fluorination: (a) G. S. Lal, G. P. Pez, R. G. Syvret, Chem. Rev. 1996, 96, 1737; (b) S. D. Taylor, C. C. Kotoris, G. Hum, Tetrahedron 1999, 55, 12431; (c) A. S. Kiselyov, Chem. Soc. Rev. 2005, 34, 1031; (d) N. Shibata, T. Ishimaru, S. Nakamura, T. Toru, J. Fluorine Chem. 2007, 128, 469.
- (a) R. E. Banks, S. N. Mohialdin-Khaffaf, G. S. Lal, I. Sharif, R. G. Syvret, J. Chem. Soc., Chem. Commun. 1992, 595; (b) R. E. Banks, J. Fluorine Chem. 1998, 87, 1; (c) R. P. Singh, J. M. Shreeve, Acc. Chem. Res. 2004, 37, 31; (c) P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent, C.-H. Wong, Angew. Chem., Int. Ed. 2005, 44, 192; (d) S. Stavber, Molecules 2011, 16, 6432.
- (a) E. Differidng, H. Ofner, Synlett 1991, 187; (b) Y. Li, Q. Zhang, Synthesis 2015, 47, 159. And See also Ref. 3a, 3d, 3e.
- K. Fukushi, S. Suzuki, T. Kamo, E. Tokunaga, Y. Sumii, T. Kagawa, K. Kawada, N. Shibata, Green Chem. 2016, 18, 1864.
- A. Harsanyi, G. Sandford, Org. Process Res. Dev. 2014, 18, 981.
- O. Lerman, S. Rozen, J. Org. Chem. 1983, 48, 724.
- T. B. Patrick, S. Nadil, J. Fluorine Chem. 1988, 39, 415.
- O. S. Tee, N. R. Iyengar, M. Paventi, J. Org. Chem. 1983, 48, 761.
- T. Umemoto, S. Fukami, G. Tomizawa, K. Harasawa, K. Kawada, K. Tomita, J. Am. Chem. Soc. 1990, 112, 8563.
- S. T. Purrington, D. L. Woodard, J. Org. Chem. 1990, 55, 3423
- H. Kamiya, M. Sato, C. Kaneko, Tetrahedron Lett. 1997, 38, 587.
- (a) R. D. Chambers, J. Hutchinson, J. Fluorine Chem. 1998, 92, 45; (b) R. D. Chambers, J. Hutchinson, J. S. Moilliet, WO 9903802 A1, 1999.
- F. Jiang, Y. Zhao, J. Hu, Org. Chem. Front. 2014, 1, 625.
- A. Harsanyi, G. Sandford, Green Chem. 2015, 17, 3000.
- (a) T. Muüh, P. Fiedler, H. Weintritt, W. Westerkamp, A. Reinecke, WO2002016304A1, 2002. (b) A. Guünther, H. Weintritt, S. Boöhm, WO2005019154A1, 2005.
- M. Braun, C. Brosch, WO2002060838A1, 2002.
- O. B. Sambueva, V. A. Yanovsky, N. F. Mingalimov, A. N. Tretyakov, O. S. Andrienko, V. I. Sachkov, Fluorine notes, 2012, 3 (82), Sam1-Sam5.
- T. Kitamura, K. Muta, J. Oyamada, Synthesis 2015, 47, 3241.
- R. Bohlmann, DE4313664, 1994.
- Transesterification was slowly observed even at room temperature. Based on these results, transesterification might also have taken place in our previous work on the fluorination of α-substituted malonates [10], although byproducts were presumably removed by column chromatography during the purification step.
Recommended for publication by Prof. Norio Shibata
Fluorine Notes, 2016, 105, 3-4
