Received: январь 2015
Fluorine Notes, 2016, 104, 3-4
ПРОСТОЙ СПОСОБ ПОЛУЧЕНИЯ ЧИСТЫХ 5,10,15,20-ТЕТРАКИС(ПЕРФТОРАЛКИЛ)ПОРФИРИНОВ
József Rábai,* Anikó Nemes, István Jalsovszky and Dénes Szabó
Institute of Chemistry, Eötvös Loránd University, P. O. Box 32, H-1518, Budapest 112, Hungary
E-mail: rabai@elte.hu
Аннотация: Предгается метод получения мезо-тетракис(перфторалкил)порфиринов без применения тонкослойной хроматографии. Данный метод включает карбинольную циклизацию, совмещенную с удалением азеотропной воды, с использованием природного глинистого кислотного катализатора. Описанная техника отделения продукта основана на существенном различии в стабильности порфиринов и олигомерных побочных продуктов в ходе окисления и их основности.
Ключевые слова: Фторные порфирины, TsOH/montmorillonite K10, 2-пиррил-карбинолы, LaCl3 темплатный эффект
Введение
Порфирины и их комлексы с металлами играют большую роль в биохимических реакциях, включая реакции окисления молекулярным кислородом [1]. Они были протестированы в качестве биомиметрических катализаторов для селективного окисления в органическолй химии [2]; фотосенсибилизаторов в фотодинамической терапии (ФДТ)[3] и красителях, увеличивающих чувствительность солнечных батарей [4]; кроме того они используется в качестве структурных элементов для супермолекулярной химии и в науке о материалах [5]; и в сенсорах [6].
Синтез электроноакцепторных порфиринов часто основывается на замещении фтора или перфторалкила на водород в молекулах исходных соединений [7]. Несмотря та то, что такие производные являются одними из наиболее активных лигандов, проверенных для активации О2, было однозначно установлено, что фторсодержащие порфирины, содержащие железо, не устойчивы в окислительных средах [8]. Несмотря на то, что данный подход не дал результата по новому фторсодержащему окислительному катализатору, это привело к разработке более широкой концепции фторной химии [9,10].
Руководствуясь статьей Therien [11], в которой раскрыта эффективная наработка мезо-(C3F7)4PorH2 лиганда, мы сосредоточились на разработке выделения целевых соединений без применения хроматографии и масштабировании метода, чтобы получить фторные порфирины в граммовых количествах. Вскоре мы применили стратегию Therien [11]: сильное разбавление для карбинольной циклизации с одновременным удалением азеотропной воды на этапе образования порфириногена, с последующим окислением 2,3-дихлор-4,5-дициано-1,4-бензохиноном (= DDQ) – но дополнили ее лишь упрощением методики по выделению продукта.
Мы подумали, что разница в основности и окислительной стабильности целевых порфиринов и их побочных олигомерных продуктов пироллинового вида могла бы быть использована для создания эффективной методики разделения. В этой статье мы раскрываем некоторые улучшенные методы, включающие проработку нестандартных методик.
Результаты и обсуждение
Синтез мезо-тетракис(перфторалкил)порифиринов [12]
Несмотря на то, что путем кислотно-каталической конденсации пиррола и альдегида получают мезо-ариловые или мезо-акриловые порфирины с хорошими выходами, следуя методикам Адлера и Лонго [13], или Lindsey [14]; Виджесекара и соавт. предоставили отчет о попытке наработать мезо-тетракис(трифторметил)порфирин и указали, что даже более жесткие условия реакции приводят к получению очень маленьких количеств данного мезо-(CF3)4PorH2 порфирина [15].
Нашей первой целью являлся мезо-тетракис(перфторгептил)порфирин (2a, R = C7F15 = Rf7). Поскольку в начале девяностых не было известно ни одной методики синтеза данного порфирина (1990’), мы выбрали метод карбинольной циклизации, описанный Куродом и его соавторами, с учётом того, что они использовали только алкил- и арил-2-пиррил-карбинолы в кипящей пропионовой кислоте. Цель добавления Zn(OAc)2 заключалась в улучшении выходов через Zn2+-ион темплатный эффект (Схема 1)[16].
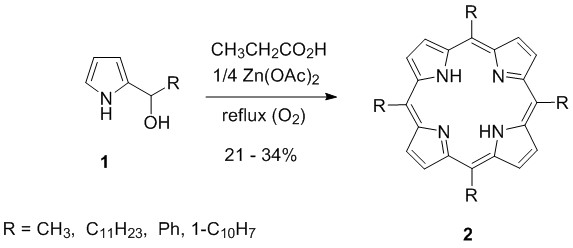
Схема 1. Синтез мезо-алкил- или арил-порфиринов на основе карбинола.
Для того, чтобы вывить стабильность данного метода синтеза месо-(перфторалкил)порфиринов мы сначала получили фтор-алкил-карбинолы (1a,b) путем восстановления с помощью NaBH4 исходных фтор-алкил-кетонов (3a,b). Фтор-алкил-карбинолы получаются с хорошим выходом, согласно основанным на литературных данных [17] методикам, которые мы оптимизировали (схема 2).
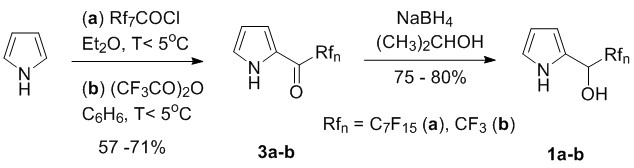
Схема 2. Улучшенный синтез фтор-алкил-(2-пиррил)-карбинолов.
Затем мы проверили 1a используя такие же условия (схема 1), но не смогли получить хоть какое-нибудь количество мезо-(Rf7)4PorH2(2a). После этого мы заменили пропионовую кислоту на фторсодержащий растворитель (R-113) и дефлегмированной 1:1 смесью 1а и TsOH*H2O. В УФ спектре не было обнаружено никакого образования 2a до тех пор пока в случайном эксперименте ½ объема растворителя не испарилась из за прекращения подачи холодной волы. Данный эксперимент при работе с (Et2O-CF3CO2H-CH3OH = UWP-1) позволил получить заметное количество целевого фторсодержащего порфирина 2a(Схема 3).
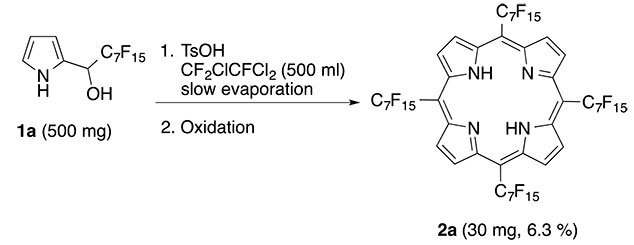
Схема 3. Непрогнозируемый синтез мезо-тетракис-(мперфторгептил)-порфирина
Причиной для образования 2a послужило удаление азеотропной воды, которое благоприятно повлияло на α-алкилирование пиррил-карбинолов при C5 благодаря образованию карбкатионов в процессе конденсации. Это полностью аналогично наблюдениям Therien, который описал эффективные синтезы (C3F7)4PorH2 [11a], по всей линии гомологов, имеющих как короткие (CF3)4PorH2 так и длинные (C7F15)4PorH2 фторуглеродные цепочки через карбинольную циклизацию [11b].
Ранее было известно, что азеотропное удаление воды увеличивает выходы октаэтил-порфирина полученного путем конденсации 3,4-диэтилпиррола и формальдегида [18], или серии мезо-тетракисарил-порфиринов, полученных путем кислотно каталитической конденсации пиррола и ароматических альдегидов [19].
Данные наблюдения в соответствии с предложенными механизмами ”4×пиррил-карбинол” [11] и ”4 пиррол + 4 алдегид” [14] методов циклизации, в которых α-пиррил-катионы и порфириногены являются универсальными промежуточными продуктами. В обоих случаях при условии отсутствия кислорода эта последовательность является обратимой и тетрамерные соединения могут подвергаться циклизации, давая порфириноген, который должен быть блокирован при максимальной концентрации с помощью DDQ. В противном случае, в медленных и обратимых условиях реакции возможно значительное увеличение побочных продуктов олигомеров. Тем не менее их формирование здесь не показано (схема 4).

Схема 4. Механизм
образования порифирина согласно Lindsey.
(Типовые
промежуточные продукты по методам Lindsey [14]
и Therien [11]
выделены синим
цветом.)
Как результат, получение максимальных выходов может быть достигнуто, если условия реакции оптимизированы для получения порфириногена, с последующим подавлением нежелательных реакций данного промежуточного соединения с помощью окислительного реагента, такого как DDQ [20]. Данное окисление превращает побочные продукты олигопиррометана (5a,b) в олигопиррометены темного цвета (6a,b) (Схема 5).
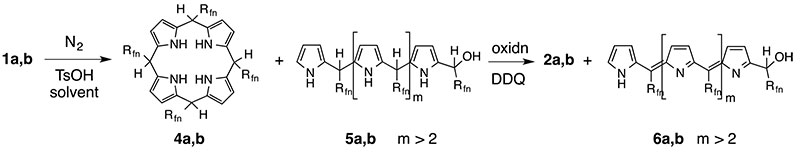
Схема 5. Двухстадийный синтез мезо-(перфторалкил)-порифиринов.
Карбинолы:1;
Порфириногены:4;
Олигопиррометаны:5;
Порфирины:2;
Олигопиррометены:6
Оптический УФ спектр мезо-тетракис(перфторалкил)порфиринов.
Интенсивность полосы Соре (Soret-band, в «синем» диапазоне волн) увеличивается, если добавляют несколько капель CF3CO2H к CF2ClCFCl2 (R-113) или к эфирному раствору таких фторсодержащих образцов. Это согласуется с образованием двойных протонированных молекул порфирина, которые демонстрируюся дикатионами типа [(Rfn)4PorH4]2+. Тем не менее, такое кислотно-щелочное равновесие может быть смещено в обратную сторону путем прибавления нескольких капель метанола, который является более сильным основанием, чем изученные порфирины.
Нами показано, что олигопиррометены 6 являются более сильными основаниями, чем соответствующие порфирины 2, соответственно позволяя формироваться соединениям поликатионного типа (7). Они окисляются намного быстрее пероксидом водорода в CF3CO2H по сравнению с первоначальными порфиринами (2), образуя более полярные продукты такие как малеимид (8) и соответствующая перфтоалканкарбоновая кислота (9) (Схема 6).
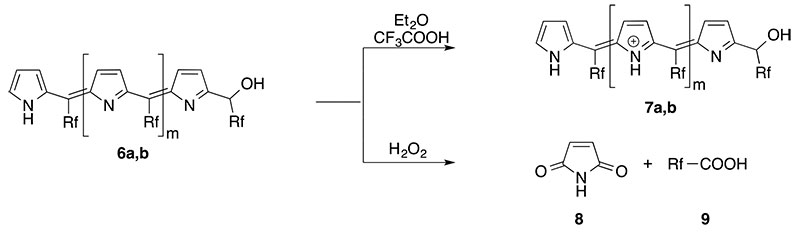
Схема 6. Кислотно-основная реакция (протонирование) и характеристики окисления олигопиррометенов 6a,b.
Данные свойства позволяют легко отделать целевые фторные порфирины 2a,b из компонентов поликатионового вида 7a,b путем осаждения с метанолом или из более полярных продуктов образовавшихся окислением олигопиррометенов. С одной стороны, добавление исбытка метанола к концентрированному эфирному/CF3CO2H раствору «сырца» смеси порфиринов и олигопирроленов будет депротонировать только порфирины и будет вызывать их коагуляцию в виде вязкообразных твердых материалов, в то время как протонированные побочные продукты остаются в растворе (UWP-1). С другой стороны, следуя двуфазной окислительной обработке смеси «сырца» порфирина и олигопирролена водным H2O2/HCO2H, простая щелочная экстракция фильтрованием через основной Al2O3 может привести к получению чистых растворов целевых фторсодержащих порфиринов (UWP-2).
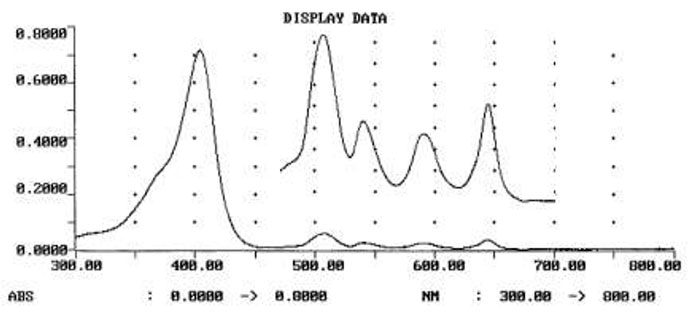
Схема 7. Видимый УФ спектр чистого 2a (в R-113): Полоса в «синей части» (Soret-band) при 404.4, Q1-Q4(вставка) при 506.4, 541.3, 588.9, 644.6 нм. Близость к нулевой отметке интенсивности поглощения при 300 нм указывает, что олигомерные побочные продукты были успешно удалены.
окислительные свойства мезо-перфторалкил-порфиринов и фторолтигопирроленов
Эксперименты с водной H2O2/CF3CO2H в процессе УФ измерений показали изначальное улучшение полосы Soret-band, но при длительном периоде наблюдается окислительная деградация всех изученных порфиринов. Данные экмперименты четко показали, что даже очень “бедные электронами” фторные порфирины и их металло-комплексы не выживают в окислительных условиях, где возможно образование пероксидных промежуточных соединений. Такие окисления обычно проводились при 20°C в двухфазной системе жидкость-жидкость, состоящей из 30% водного H2O2/CF3CO2H – R-113 пары растворителей. Позже озоноразрушающий R-113 заменили бензотрифторидом (БТФ) или CH2Cl2 в зависимости от растворимости образцов «сырца» F- порфиринов. Чтобы замедлить окисление порфиринов в образцах «сырца» для препаративного разделения, CF3CO2H заменили на HCO2H. Потеряв лишь небольшую часть порфиринов, все побочные продукты были превращены в более полярные соединения используя системы: водный H2O2-HCO2H/ БТФ, толуол или CH2Cl2 , Эти соединения легко абсорбируются на щелочном оксиде алюминия или могут удаляться путем водной NaOH-экстракции.
метод A:
Мы использовали катализированную кислотой карбинольную циклизацию в слабом растворе (2.5-5 × 10-3 M) в бензоле одновременно удаляя воду путем отгонки азеотропного растворителя, как сообщено Therien с соавторами, с последующей обработкой порфириногена DDQ, но используя двухфазную жидкость-жидкость среду окисления и кристаллизацию для улучшенного выделения продукта. Мы получили порфирина 2a [230 мг (7.1%)] в чистом виде с температурой плавления = 248-249oC. У нас на руках были только выходы (34%) сырца 2a как и в сообщениях.
Метод B
Метод карбинольной циклизации при сильном разбавлении толуолом (2.2 × 10-3 M) в присутствии ХЛОРИДА ЛАНТАНА (III) проводили с помощью серной кислоты на кремнеземном носителе, что помогало конденсации, с одновременным удалением воды путем азеотропной отгонки растворителя, за которой следовала обработка с помощью DDQ промежуточного порфириногена. При этом применяли двухфазную жидкость-жидкость среду окисления и кристаллизацию для улучшенного выделения продукта. Мы получили порфирин 2a [900 мг (18.8%)] в чистом виде с Тпл = 248-249oC. Успех новой методики может быть связан с образованием координационного комплекса между оксофильными La3+ катионами и исходными карбинолами (темлантный эффект). Без LaCl3 не наблюдалось никакого заметного образования порфирина, но происходило полное разложение карбинола в момент введения H2SO4*SiO2.
метод_C:
Кислота
нанесенная на глинозем
[21] катализировала
карбинольную циклизацию при высоком разбавлении толуолом с
одновременным удалением воды через азеотропную отгонку растворителя,
за
которой следовала обработка с помощью DDQ промежуточного
порфириногена, с
последующим применением двухфазной жидкость-жидкость среды окисления
и дальнейшей кристаллизацией для улучшенного выделения продукта..
Образцы
чистых (C7F15)4porH2 и
(CF3)4porH2 с
четкой точкой плавления были легко получены в граммовых количествах с
выходами 15 или 25%, соответственно.
Экспериментальная часть
Пиррил-карбинолы были получены путем восстановления с помощью NaBH4 предшествующих кетонов, как показано ниже, и их хранили при 0°C до момента использования. Растворители и реактивы были куплены в компаниях Sigma-Aldrich и Molar. 1H-, 13C- и ЯМР спектры 19F были сняты на Bruker Avance 250 с использованием 5 мм инверсной 1H/13C/31P/19F измерительной головки при комнатной температуре. Химические сдвиги (δ) даны в миллионных долях (м.д.) относительно внутреннего стандарта ТМС (δ=0.00 для 1H, δ=0.00 для 13C) и CFCl3 в качестве внешнего стандарта (δ=0.00 для 19F). Температуры плавления определялись приборе для определения температуры плавления для микроколичеств вещества марки «Boetius», и не учитывали поправки. За реакциями следили с помощью тонкослойной хроматографии (SiO2 и Al2O3 подложки), УФ спектроскопии (Carry) и газовой хроматографии (Hewlett-Packard 5890 Series II, PONA [сшитая метисиликоновая смола] 50 м x 0.2 мм x 0.5 мм колонка, H2 газоноситель, пламенно ионизационный детектор; программа : 120 °C, 5 мин, 10 °C/мин, 250 °C, 5 мин; инжектор: 250 °C, детектор : 280 °C).
2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-Пентадекафтор-1-(2’-пирролил)-1-октанон (3a)
К раствору 2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-пентадекафтороктаноил хлорида (15.15 г, 0.035 молей) в эфире (35 мл) по каплям добавляли раствор пиррола (2.41 г, 0.036 молей) в эфире (30 мл) при перемешивании в течение 30 минут. После прибавления смесь перемешивали еще 2 часа при комнатной температуре. Реакционную смесь отмывали 5% NaHCO3 раствором (2 x 50 мл) и водой, затем сушили над сульфатом натрия. Растворитель удаляли, затем сырец кристаллизовали из пентана и получали 8.2 г (57 %) чистого продукта. Температура плавления 48-49 °C.
ЯМР спектр 1H (250 МГц CDCl3) δ (м.д.): 6.43 (1H, м), 7.28 (2H, м), 9.98 (1H, с).
2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-Пентадекафтор-1-(2’-пирролил)-1-октанол (1a)
К охлажденному водой со льдом и перемешиваемому раствору 2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-пентадекафтор-1-(2’-пирролил)-1-октанона (9.27 г, 0.020 мол) в смеси растворителей (130 мл тетрагидрофурана и 60 мл метанола) постепенно добавляли NaBH4 (1.76 г, 0.046 мол) маленькими порциями в течение 30 минут. После перемешивания в течение 2 часов при комнатной температуре добавляли эфир (50 мл) и раствор промывали насыщенным NaHCO3 (50 мл). Органическую фазу сушили над Na2SO4. После удаления растворителя осадок кристаллизовали из толуола (40 мл) и получали 7.0 г (75%) чистого кристаллического продукта, Тпл = 93-95 °C.
ЯМР спектр 1H (250 МГц CDCl3) δ (м.д.): 2.95 (1H,с), 5.00 (1H, м), 6.23 (1H, м), 6.31 (1H, с), 6.83 (1H, м) 8.59 (1H, с).
2-Трифторацетил-пиррол (3b)
К охлажденному раствору трифторуксусного ангидрида (46 мл, 0.325 молей) в эфире (400 мл) по каплям добавляли раствор пиррола (19.0 г, 0.283 молей) в эфире (55 мл) при перемешивании в течение 2.5 часов. После добавления смесь перемешивали при 0°C еще 4 часа. Реакционную смесь промывали водой, затем сушили (Na2SO4). Растворитель удаляли, затем осадок перегоняли с водяным паром. Дистиллят охлаждали до 0 °C, образовующееся кристаллическое вещество отфильтровывали, промывали холодной водой и сушили с полученим 32.7 г (71 %) продукта. Температура плавления 38-40°C (в работе [17] температура плавления 46-47 °C/ получено вакуумной возгонкой).
ЯМР спектр 1H (250 МГц CDCl3) δ (м.д.): 6.43 (1H, т), 7.26 (1H, с), 7.31(1H, с), 10.18 (1H, с)
2,2,2-Трифтор-1-(2’-пирролил)-1-этанол (1b)
При перемешивании к охлажденному раствору 2-трифторацетил пиррола (25 г, 0.153 молей) в метаноле (100 мл) осторожно добавляли NaBH4 (12.5 г, 0.33 молей) маленькими порциями в течение 3 часов. После перемешивания в течение 1 часа добавляли насыщенный NaHCO3 (50 мл) и смесь экстрагировали с эфиром (5 x 50 мл), затем комбинированную органическую фазу промывали насыщенным NaHCO3, выдерживали в соляном растворе, и сушили над Na2SO4. После удаления растворителя осадок отгоняли при вакууме с полученим 20.3 г (80.4%) бесцветного масла с температурой кипения 112-120°C/18 мм. рт. ст.; которое превращается в твердое белое вещество при комнатной температуре, температура кипения 45-47°C.
ЯМР 1H (250 МГц CDCl3) δ (м.д.): 2.95 (1H, с), 5.01 (1H, к), 6.22 (1H, м), 6.31 (1H, с), 6.83 (1H, м), 8.59 (1H,с).
Монтмориллонит K10/TsOH*H2O реактив [Глина/TsOH (4:1 вес./вес.)]
Монтмориллонит K10 (50 г) перемешивали с раствором моногидрата п-толуолсульфоновой кислоты (10 г) в дистиллированной воде (150 мл). Смесь гомогенезировали испарением большей части воды с использованием роторного испарителя, затем сушили в вакууме над P2O5 3 дня. Количественный выход (60 г).
H2SO4/SiO2 реактив[10% вес./вес.H2SO4/SiO2]
Серную кислоту (5.00 г, 96%) перемешивали в колбе с SiO2 (45 г, Merck Kieselgel 40, 35-70 меш). Наблюдали некоторое нагревание. Колбу закупоривали и встряхивали руками до тех пор, пока не наблюдали однородную массу реактива. Затем нагревали до 80°C и выдерживали под вакуумом (16 мм.рт.ст.) в течение 5 минут. После этого охлаждали до комнатной температуры и хранили в закупоренной колбе. Выход : ~1.0 mmol H2SO4/г твердого вещества.
Мезо-Тетракис(перфторгептил)порфирин (2a)
Вышеуказанное соединение было приготовлено по методу A с использованием 5×10-3 M карбинола в присутствии 2.5×10-3 M TsOH катализатора. 3.40 г (7.31 mmol) C7F15-(α-пиррил)-карбинол (1a); 1.5 л бензола и 0.72 г (3.78 ммол) TsOH*H2O перемешивали над N2 и ½ объема растворителя отгоняли. DDQ (1.73 г, 7.62 ммол) добавляли и выдерживали реакционную массу под обратным холодильником в течение часа. После этого реакционную массу отфильтровывали и выпаривали до сухого состояния. Осадок был обработан смесью CF2ClCFCl2 (700 мл), HCO2H (350 мл) и 30% водный H2O2 (35 мл) и выдерживали реакционную массу под обратным холодильнком в течение 5 часов. Затем смесь растворяли в воде (350 мл), фазы разделяли и слой CF2ClCFCl2 (R-113) промывали 0.2 M NaOH (5×700 mL). Сушили над Na2SO4 и фильтрат выпаривали. Далее проводли перекристализацию из CF3C6H5/CH3OH с полученим 230 мг (0.129 ммол, 7.1%) 2a с Тпл = 248-249°C.
Данные спектроскопии согласуются с данными из работы [11b]. ЯМР спектр 1H (250 МГц, CDCl3, CF2ClCFCl2): δ 9.54 (с, 8 H), -2.25 (с 2 H); ЯМР спектр 19F (CDCl3): δ-80.7, (br, 2 F), -82.3, (br, 3 F), -115.3, (br, 2 F), -121.7, (br, 2 F), -122.1, (br, 2 F), -122.8, (br, 2 F), -126.7, (br, 2 F); УФ спектр (CF2ClCFCl2) (Rf7)4PorH2: λmax = 404 нм (εmax= 143,760 Soret); [(Rf7)4porH4]2+: λmax = 423 нм (образование дикатионов приводит к сдвигу на 19 нм в «красную» область).
мезо-Тетракис(перфторгептил)порфин (2a)
Вышеуказанное соединение было приготовлено по методу B с использованием 2×10-3 M карбинола и 1×10-3 M LaCl3*7H2O заготовки в толуоле. Смесь 1a (5.00 г, 10.75 ммоль), LaCl3*7H2O (2.00 г, 5.39 ммоль) и толуол (5 Л) перемешивали над N2, нагревали электроколбонагревателем до появления обратного потока, и отгоняли около 1 литра толуола (~30 мин). Обнаруживали бледно фиолетовый цвет. Добавляли еще 1 Л толуола вместе с 10%H2SO4/SiO2 (2.50 г; = 250 мг H2SO4, 2.55 моль), перемешивали и выдерживали с обратным холодильником. Затем толуол (500 мл) отгоняли в течении 30 минут чтобы появилось потемнение цвета. Затем добавили DDQ (2.50 г, 11.0 моль) и смесь выдерживали при обратном холодильнике в течение часа. Пока смесь была горячей, ее отфильтровывали и упаривали (Роторный испаритель). Полученное твердое вещество отмывали CH3OH (200 мл) и сушили до получения 3.10 г сырца. Растворяли в CF2ClCFCl2 (500 мл) и перемешивали со смесью HCO2H (250 мл) и 30% водной H2O2 (25 мл) в течение 12 часов. Отделяли нижнюю фазу и промывали 85% HCO2H (3×100 мл), затем водой (3×200 мл), 1M KOH (3×200 мл) и в завершении рассолом. Сушили над Na2SO4, отфильтровывали и извлекали растворитель при атмосферной отгонке для получение обогащенной твердой фракции порфирина: 1.76 г (~36%) после осушки при 80°C/16 мм. рт. ст. Дважды перекристаллизовывали из смеси CF3C6H5 (12 мл) и CH3OH (5 мл) и получали 900 мг (18.8 %) аналитически чистого (Rf7)4PorH2 в качестве кристаллов темно фиолетового цвета; температура плавления = 248-249°C. УФ спектр (R-113): Полоса Soret 404 нм (ε = 143,760).
мезо-Тетракис(перфторгептил)порфирин (2a)
Вышеуказанное соединение было приготовлено по методу C с использованием 2×10-3 M карбинола и 5×10-4 M TsOH*/K10 в толуоле. Смесь толуола (5.0 Л) и 1:5 (массовые доли) TsOH*H2O/Montmorillonite K10 (600 мг; 0.53 ммоль TsOH) и 1a (5.00 г, 10.75 ммоль) перемешивали и нагревали в медленном потоке N2 затем отгоняли около 1 литра толуола в течение часа. Полученную смесь обрабабатывали DDQ (3.50 г, 15.4 ммоль) и кипятили с перемешиванием еще час. Горячую реакционную смесь отфильтровывали через фильтровальную бумагу. Данный фильтрат толуола давал темный осадок при 20°C, но значительное количество порфирина оставалось в растворе. Затем раствор упаривали в вакууме (Роторный испаритель) и осадок выщелачивали с CH3OH (400 мл), чтобы удалить оставшиеся нефторированные побочные продукты (хинон/гидрохинон). Фторсодержащий осадок и остатки объединяли растворением в CF2ClCFCl2 (R-113, 500 мл). Экстракт сырца фторсодержащего порфирина и олигомерных пиррометенов в R-113 перемешивали со смесью HCO2H (250 мл) и 30% водной H2O2 (25 мл) в течение 24 часов. После этого двухфазную жидкостную систему обрабатывали следующим образом: отделенный слой CF2ClCFCl2 последовательно промывали 85% HCO2H (100 мл), затем H2O (2×100 мл), 1M KOH (3×200 мл) и рассолом (2×50 мл). Раствор сушили (Na2SO4), фильтровали и упаривали, чтобы получить ~2.5 г обогащенного продукта. Повторная (2×) перекристаллизация полученного из 3:2 об./об. смеси CF3C6H5 и CH3OH давала кристаллы темно фиолетового цвета, 1.208 г (0.6778 ммоль, 25.2%) (Rf7)4PorH2, с Тпл = 246-248°C. УФ спектр (R-113): полоса Soret 404 нм (ε = 143,760).
мезо-Тетракис(трифторметил)порфирин (2b)
Вышеуказанное соединение было приготовлено по методу C с использованием 6×10-3 M CF3-карбинола и 3×10-4 M TsOH*momoK10 в толуоле. Смесь толуола (5.0 л) и 1:5 вес./вес. TsOH*H2O/Montmorillonite K10 (1.60 g; 1.40 ммоль TsOH) и 1b (5.00 г, 30.3 ммол) перемешивали и нагревали в слабом потоке N2 , затем 1 л толуола отгоняли в течение часа. Добавляли DDQ (6.0 г, 26.4 ммоль) к смеси и кипятили в течение 1 часа. Затем добавляли основной (Brockman I) Al2O3 (250 г) и смесь отфильтровывали пока она было горячей, фильтрат упаривали в вакууме (Роторный испаритель). Осадок растворяли в CF2ClCFCl2 (500 мл) а затем перемешивали с раствором 30% H2O2 (50 мл) в 98% HCO2H (500 мл) при комнатной температуре в течение 24 часов. Жидкую фазу отделяли, затем слой CF2ClCFCl2 промывали H2O (2×100 мл), 1M KOH (3×200 мл) и рассолом (100 мл). Раствор CF2ClCFCl2 сушили (Na2SO4), отфильтровывали и упаривали, чтобы получить ~2 г обогащённого продукта. Перекристаллизовывали из CF3C6H5-CH3OH (9:1 вес./вс.), а затем из 2-пропанола (25 мл/100 мг), чтобы получить 670 мг (1.15 mM, 15.2%) аналитически чистых темно синих блестящих кристаллов; Тпл= 192-196°C.
Данные спектроскопии согласовывались с результатамие [11b]. ЯМР 1H (250 МГц, CDCl3): δ 9.60 (с, 8H), -2.08 (с, 2H); ЯМР спектр 19F (CDCl3): δ -38.41 (с, 3F). Vis (CH2Cl2): УФ полоса Soret 403 нм (ε = 120, 226), 510, 545, 593, 649.
благодарности
Данное исследование было выполнено при поддержке Венгерского Научно-Исследовательского фонда (OTKA K 062191 “Sustainable fluorous chemistry”)
Список литературы
- Enikolopyan, N. S.; Bogdanova, K. A.; Askarov, K. A.;Metal Complexes of Porphine and Azaporphine Compounds as Catalysts of Reactions Involving Oxidation by Molecular Oxygen; Russian Chemical Reviews, 1983, 52, 13-26; Translated from Uspekhi Khimii, 1983, 52, 20-42.
- Mansuy, D., Biomimetic catalysts for selective oxidation in organic chemistry, Pure & Appl. Chem., 1990,62, 741-746.
- (a) Li,G.;Chen, Y.; Missert, J. R.; Rungta, A.; Dougherty, T. J.;Grossman, Z. D.; Pandey, R. K.;Application of Ruppert’s reagent in preparing novel perfluorinated porphyrins, chlorins and bacteriochlorins, J. Chem. Soc.,Perkin Trans. 1, 1999, 1785–1787. (b) Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey R. K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy, Chem. Soc. Rev. 2011, 40, 340-362.
- (a) Li, L.-L.; Diau, E. W.-G. Porphyrin-sensitized solar cells, Chem. Soc. Rev. 2013, 42, 291-304; (b) Urbani, M.; Grätzel, M.; Nazeeruddin, M. K.; Torres, T. Meso-Substituted Porphyrins for Dye-Sensitized Solar Cells, Chem. Rev. 2014, 114, 12330-12396.
- Briza, T.; Kaplánek, R.; M; Havlik, M.; Dolensky, B.; Kejik, Z.; Martasek, P.; Král, V., Synthesis of Highly Functionalized Fluorinated Porphyrins; Supramolecular Chemistry, 2008, 20 (3), 237–242.
- (a) Král, V.; Králová, J.; Kaplánek, R.; Bríza, T.; Martásek, P., Quo vadis porphyrin chemistry?Physiol. Res. 2006, 55 (Suppl. 2): S3-S26. (b) Chen, L. D.; Zou, X. U.; Buhlmann, P. Anal Chem. 2012, 84, 9192-9198.
- (a) Belyaeva, E. V.; Sigan, A. L.; Druzhinina, I. E.; Ikonnikov, N. S.; Chkanikov, N. D.; A method of introducing fluorinated substituents in porphyrin structure by nucleophilic substitution of fluorine in meso-tetrakis(pentafluorophenyl)porphyrin and pentafluorobenzaldehyde with polyfluoroaliphatic alcohols. Fluorine notes, Vol. 5(102) 2015; (b)Aggarwal, A.; Singh, S.; Samson, J.; Drain, C. M. Adaptive Organic Nanoparticles of a Teflon-Coated Iron (III) Porphyrin Catalytically Activate Dioxygen for Cyclohexene Oxidation. Macromol. Rapid Commun. 2012, 33, 1220−1226.
- (a) Moore, K. T.;Horváth, I. T.; Therien, M. J. High-Pressure NMR Studies of (Porphinato) iron-Catalyzed Isobutane Oxidation Utilizing Dioxygen as the Stoichiometric Oxidant; J. Am. Chem. Soc.,1997,119 , 1791–1792; (b) Moore, K. T.; Horváth, I. T.;Therien, M. J., Mechanistic studies of (porphinato) iron-catalyzed isobutane oxidation. Comparative studies of three classes of electron-deficient porphyrin catalysts.Inorganic Chemistry, 2000,39 (15), 3125-3139.
- (a) Horváth, I. T.,Rábai, J. Facile Catalyst Separation without Water: Fluorous Biphase Hydroformylation of Olefins. Science1994, 266, 72-75; (b)Fluorous Multiphase Catalyst or Reagent Systems for Environmentally Friendly Oxidation or Hydroformylation or Extraction Processes. Horvath, I. T.; Rabai, J. (Exxon Research and Engineering Co., USA). Eur. Pat. Appl. (1995), 11 pp. EP 633062 A1 19950111; (c) Metal-Fluorinated and Metal-Perfluorinated Complexes as Catalysts and Extractants for Multiphase Systems. Horváth, I. T.; Rábai, J. (Exxon Research and Engineering Co., USA). U.S. (1999), 8 pp., US 5981422 A 19991109.
- (a) Horváth, I.T. A personal view of the history of fluorous chemistry. Chapter 2; pp. 5-10. In: Gladysz, J. A.; Curran, D.P.; Horvath, I. T. (Eds): Handbook of Fluorous Chemistry, Wiley-VCH, 2004, Weinheim, Germany; (b) Fluorous Chemistry, Volume Editor: Istvan T. Horvath, Vol. 308, 2012; in: Topics in Current Chemistry, Springer-Verlag Berlin Heidelberg 2012.
- (a) DiMagno, S. D.; Williams, R. A.; Therien, M. J., Facile Synthesis of meso-Tetrakis(perfluoroalkyl)porphyrins: Spectroscopic Properties and X-ray Crystal Structure of Highly Electron-Deficient 5,10,15,20-Tetrakis (heptafluoropropyl)porphyrin.J. Org. Chem., 1994, 59, 6943-6948; (b) Goll, J. G.; Moore, K. T.; Ghosh, A.; Therien, M. J., Synthesis, Structure, Electronic Spectroscopy, Photophysics, Electrochemistry, and X-ray Photoelectron Spectroscopy of Highly-Electron-Deficient [5,10,15,20-Tetrakis (perfluoroalkyl)porphinato]zinc(II) Complexes and Their Free Base Derivatives. J. Am. Chem. Soc., 1996, 118, 8344-8354.
- For an overview of synthetic startegies and methods for porphyrins, see: (a) Wijesekera, T. P.; Dolphin, D. ’Synthetic Aspects of Porphyrin and Metalloporphyrin Chemistry’ In: Roger A. Sheldon (Editor), Metalloporphyrins in Catalytic Oxidations, Marcell Dekker, New York, 1994, Chapter 7, pp. 193-239.
- Adler, A. D.; Longo, F. R.; J. Finarelli, J.; Goldmacher, J.; Assour, J.; L. Korsakoff. L.; A simplified synthesis for meso-tetraphenylporphine. J. Org. Chern.1967,32, 476-476..
- Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M., Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987,52, 827-836.
- (a) Wijesekera, T. P.; 5-Perfluoroalkyldipyrromethanes and Porphyrins Derived Therefrom; Can. J. Chem.1996, 74, 1868-1871; (b) Wijesekera, T. P.; Lyons, J. E.; Ellis, P. E., Jr.; Bhinde, M. V., Pophyrins and Metal Complexes Thereof Having Haloalkyl Side Chains,US 5,608,054 (1977). (c) Wijesekera, T. P.; Lyons, J. E.; Ellis, P. E., Jr..Porphyrins And Their SynthesI From Dipyrromethanes And Aldehydes,US 5,760,217 (1998).
- Kuroda, Y.; Murase, H.; Suzuki, Y.; Ogoshi, H., A new route for meso-substituted porphyrin, Tetrahedron Lett. 1989,30, 2411-2412.
- Cooper, W. D.; Synthesis 2-Trifluoroacetylpyrole; J. Org. Chem., 1958, 23, 1382-1382.
- Sessler, J. L.; Mozaffari, A.; Johnson, M. R., 3,4-Diethylpyrrole and 2,3,7,8,12,13,17,18-Octaethylporphyrin, Org. Synth. 1992, 70, 68-78.
- А. С.Сейкин, О. И. Койфан, Б. Д. Березин, Уличшенный Метод Замещенных Тетрафенилпорфинов,Химия Гетероциклических Соединений,1986, No. 6, c. 798-801.
- (a) DiMagno, S. D.; Williams, R. A.; Therien, M. J., Facile Synthesis of meso-Tetrakis(perfluoro-Alkyl)-porphyrins: Spectroscopic Properties and X-ray Crystal Structure of Highly Electron-Deficient 5,10,15,20-Tetrakis (heptafluoropropyl)porphyrin.J. Org. Chem., 1994, 59, 6943-6948; (b) Therien, M. J., DiMagno, S. D.; Pyrrolic Compounds, US 5,817,830 (1998); (c) Therien, M. J., DiMagno, S. D.; Electron-deficient Porphyrins and Processes and Intermediates for Preparing the Same, US 5,856,515 (1999); (d) Moore, K. T.; Fletcher, J. T.; Therien, M. J.; Syntheses, NMR and EPR Spectroscopy, Electrochemical Properties, and Structural Studies of [5,10,15,20-Tetrakis(perfluoroalkyl) porphinato]iron(II) and -iron(III) Complexes. J. Am. Chem. Soc. 1999, 121, 5196-5209.
- Onaka, M.; Shinoda, T.; Izumi, Y., Nolen, E. Porphyrin Synthesis in Clay Nonospaces. Chemistry Letters, 1993, 117-120.
Статья рекомендована к публикации членом редколлегии В.В. Корниловым
Fluorine Notes, 2016, 104, 3-4
