Received: January, 2016
DOI 10.17677/fn20714807.2016.01.02
Fluorine Notes, 2016, 104, 3-4
Facile preparation of pure 5,10,15,20-tetrakis(perfluoroalkyl)porphyrins
József Rábai,* Anikó Nemes, István Jalsovszky and Dénes Szabó
Institute of Chemistry, Eötvös Loránd University, P. O. Box 32, H-1518, Budapest 112, Hungary
E-mail: rabai@elte.hu
Abstract: A chromatography-free method for the preparation of meso-tetrakis(perfluoroalkyl)porphyrins is suggested. This method involves carbinol cyclization coupled with azeotropic water removal using clay supported acid catalyst. Product isolation techniques disclosed here are based on inherent differences in stability of porphyrins and oligomeric side products toward oxidation and in their basicity.
Keywords: fluorous porphyrins, TsOH/montmorillonite K10, 2-pyrryl-carbinols, LaCl3 template effect
Introduction
Porphyrins and their metal complexes play great role in biochemical reactions involving oxidation reactions by molecular oxygen [1]. They are tested as biomimetic catalysts for selective oxidation in organic chemistry [2]; photosensitizes in photodynamic therapy (PDT)[3] and in dye sensitized solar cells [4]; or used as building blocks for supramolecular chemistry and materials science [5]; and in sensors based on their inherent molecular recognition properties [6].
Synthesis of electron-deficient porphyrins is often based on fluorine or perfluoroalkyl-substitution for hydrogens in the molecules of the parent compounds [7]. Although such derivatives are among the most robust ligands tested for O2 activation, it was clearly established that fluorous iron prophyrins are not stable in oxidative environments [8]. Although this approach has not resulted in new fluorous oxidation catalyst, it led to the development of a much broader concept of fluorous chemistry [9,10].
Motivated by the Therien paper [11], which disclosed the effective preparation of meso-(C3F7)4PorH2 ligand we turned our attention to develop chromatography-free isolations and scaling up batch sizes that could provide fluorous porphyrins at the gram scale. We soon applied the strategy of Therien [12] – using high dilution for carbinol cyclization with azeotropic water removal in the porphyrinogen formation step, which is followed by oxidation with 2,3-dichloro-4,5-dicyano-1,4-benzoquinone (= DDQ) – but supplemented it with simple isolation protocols.
We thought that the differences in the basicity and oxidative stability of target porphyrins and their oligopyrrolene type side products could be exploited for divising effective separation protocols. Here we disclose some improved methods involving unusual work-up protocols (UWP’s) (cf. later).
Results and Discussion
Synthesis of meso-tetrakis(perfluoroalkyl)porphyrins [12]
Although acid-catalyzed condensation of pyrrole and an aldehyde produces meso-aryl or meso-alkyl porphyrins in good yields, following the Adler and Longo [13], or the Lindsey protocols [14]; Vijesekara et al. reported that attempts to prepare meso-tetrakis(trifluoromethyl)porphyrin, even under harsher reaction conditions, lead only to very small quantities of this meso-(CF3)4PorH2 porphyrin [15].
Our first target was meso-tetrakis(perfluoroheptyl)porhyrin (2a, R = C7F15 = Rf7). Since no procedure was known for the synthesis of this porphyrin at the early nineties (1990’), we choosed the carbinol cyclization method of Kuroda et al., since they used only alkyl- and aryl-2-pyrryl-carbinols in boiling propionic acid. The role of the added Zn(OAc)2is believed to improve yields via Zn2+-ion template effect (Scheme 1)[16].
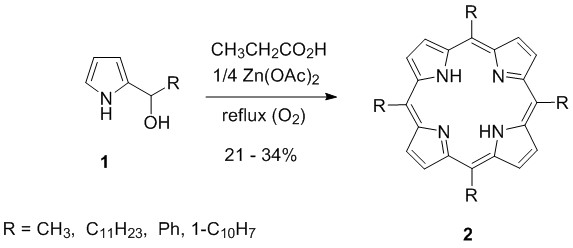
Scheme 1. Carbinol based synthesis of meso-alkyl- or aryl-porphyrins.
To reveal the suitability of this method for the synthesis of meso-(perfluoroalkyl)porphyrins we made first F-alkyl-carbinols (1a,b) by NaBH4 reduction of precursor F-alkyl-pyrryl-ketones (3a,b), which could be prepared in good yield by our optimized procedures based on literature precedences (Scheme 2) [17].
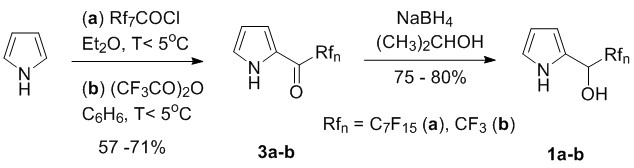
Scheme 2. Improved synthesis of F-alkyl-(2-pyrryl)-carbinols.
Then we tested 1a using same conditions (Scheme 1), but it failed to afford any meso-(Rf7)4PorH2(2a). Next we changed propionic acid to a fluorous solvent (R-113) and refluxed a 1:1 mixture of 1a and TsOH*H2O. No formation of 2a was detected by UV-V is until in a serendipitious experemint ½ volume of the solvent escaped due to a brake of cooling water supply. This test experiment on work up (Et2O-CF3CO2H-CH3OH = UWP-1) gave noticable amount of the target fluorous porphyrin 2a (Scheme 3).
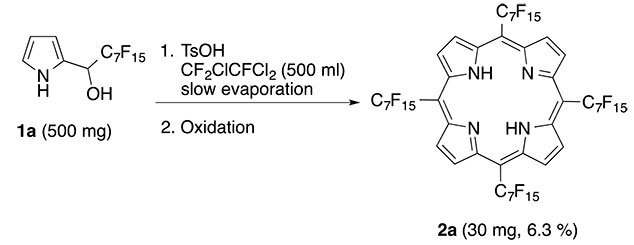
Scheme 3. Serendipitious synthesis of meso-tetrakis-(perfluoroheptyl)-porphyrin.
The reason for the formation of 2a was the azeotropic water removal, which affected beneficially the α-alkylation of pyrryl-carbinols at C5 by the carbocations formed during condensation. This is in complete analogy to the observations of Therien, who disclosed effective synthesis of (C3F7)4PorH2 [11a], along with that of shorter (CF3)4PorH2 or longer F-chain (C7F15)4PorH2 homologues via carbinol cyclization [11b].
There was known earlier that azeotropic removal of water increases the yields of octaethyl-porphyrin prepared by condensation of 3,4-diethylpyrrole and formaldehyde [18], or that of a series of meso-tetraaryl-porphyrins obtained by acid catalysed condensation of pyrrole and aromatic aldehydes [19].
These observations are in accordance with the proposed mechanisms of the ”4×pyrryl-carbinol” [11] and of the ”4 pyrrole + 4 aldehyde” [14] cyclization methods, where α-pyrryl-cations and porphyrinogen are common intermediates. In both cases under anaerobic condition this sequence is reversible and the tetrameric species may undergo cyclization to afford porphyrinogen which should be quenched at their maximal concentration by DDQ. Otherwise, under inert and reversible reaction conditions a significant increase of oligomeric side products is possible. However, their formation is not shown here (Scheme 4).

Scheme
4. Mechanism of the porphyrin
formation according to Lindsey.
(Common
intermediates of the Lindsey[14] and Therien [11] methods are shown
in blue color.)
As a consequence of the formation mechanism optimal yields could be achieved if the reaction conditions are optimized for porphyrinogen formation, then this intermediate is quenched with an appropriate oxidazing agent, such as DDQ [20]. This oxidation converts the oligopyrromethane (5a,b) side products into dark coloured oligopyrromethenes (6a,b) (Scheme 5).
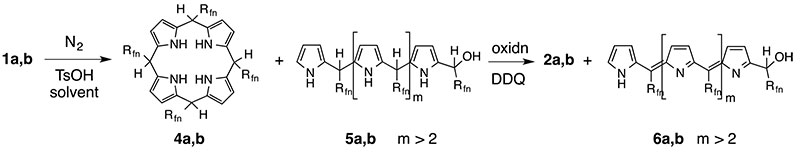
Scheme
5. Two step synthesis of meso-(perfluoroalkyl)-porphyrins.
Carbinols:1;
Porphyrinogens:4;
Oligopyrromethanes:5;
Porphyrins:2; Oligopyrromethenes:6
UV-Vis Spectroscopy of meso-Tetrakis(Perfluoroalkyl)Porphyrins.
The intensity of the Soret-band increases if a few drops of CF3CO2H is added to the CF2ClCFCl2 = R-113/or ether solution of such fluorous samples. This is in line with the formation of double protonated porphyrin molecules, which are exposing [(Rfn)4PorH4]2+ type dications. However, this acid-base equilibrium can be reversed by adding a few drops of CH3OH, a stronger base than the porhyrins studied.
We have shown that oligopyrromethenes 6 are more basic than the respective porphyrines 2, thus allowing the formation of polycation type species (7) and they are oxidized much faster with hydrogen peroxide in CF3CO2H than the parent porhyrins (2) to afford more polar product such as maleimid (8) and the corresponding perfluoroalkane carboxylic acid (9) (Scheme 6).
Scheme 6. Acid-base reaction (protonation) and oxidation characteristics of oligopyrromethenes6a,b.
These characteristic properties allow the easy separation of target fluorous porphyrins 2a,b from polycation type species 7a,b by precipitation with methanol or from more polar products formed by the oxidation of oligopyrromethenes. At one hand, the addition of excess methanol to a concentrated ethereal/CF3CO2H solution of a crude mixture of porphyrins and oligopyrrolenes will deprotonate only the porphyrin content and induce their precipitation as waxy solid materials, while the protonated side products remain in solution (UWP-1).On the other hand, following atwo phase oxidative treatment of the crude porhyrin and oligopyrrolene mixtures with aq-H2O2/HCO2H, a simple alkaline extraction of filtration through basic Al2O3 may result in the formation of pure solutions of the target fluorous porphyrines (UWP-2).
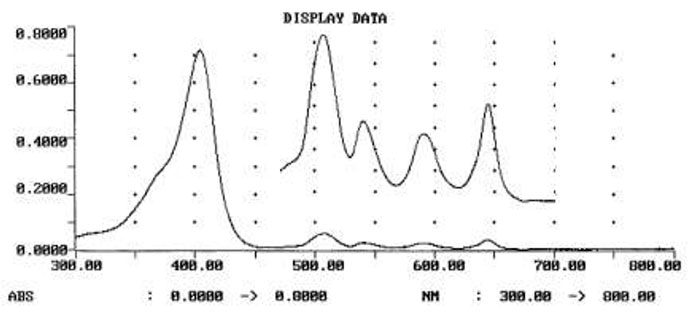
Scheme 7. UV-Vis of pure 2a(in R-113): Soret at 404.4, Q1-Q4(insert) at 506.4, 541.3, 588.9, 644.6 nm. Near to baseline absorbance at 300 nm indicates that oligomeric side products were successfully removed.
Oxidation properties of mezo-perfluoroalkyl-porhyrins and F-oligopyrrolenes
Fainting experiments with aq-H2O2/CF3CO2H during UV-Vis measurements showed an initial sharpening of the Soret-band, but on the long run resulted in the complete oxidative degradation of all the porphyrins studied. These experiments clearly indicated that even highly ’electronpoor’ fluorous porphyrins and their metal complexes does not survive oxidative conditions where formation of peroxide intermediates is possible. Such oxidations were performed at 20°C usually in a two liquid-liquid phase system, consisting of 30% aq-H2O2/CF3CO2H – R-113 solvent pair. Later the ozone depleting R-113 solvent was replaced with benzotrifluoride (BTF) or CH2Cl2 depending on solubility pattern of crude F-porphyrins. To slow down the oxidation of the porphyrin content of the crude samples that allow preparative scale separations CF3CO2H was changed to HCO2H. With the expense of sacrifying only a small part of porphyrins, all side products were converted to more polar compounds – using aq-H2O2-HCO2H/BTF, toluene or CH2Cl2 systems – which are easily adsorbed on basic alumina or could partly be removed by aq-NaOH-extraction.
Method_A:
We
used acid catalyzed carbinol cyclization at high dilution (2.5-5 ×
10-3 M) in benzene with simultanous water removal via azeotropic solvent
distillation as reported by Therien et al., followed with DDQ
quenching of the porphyrinogen, but using liquid-liquid two phase
oxidation and crystallization for improved product isolation. We
obtained porphyrin 2a [230 mg (7.1%)] in
pure state with mp = 248-249°C.
In our hands only the crude yields of 2a were as high as the
reported 34%.
Method_B:
Lantane(III)-chloride
assisted carbinol cyclization method at high dilution in toluene(2.2
× 10-3 M) was executed by silica supported sulfuric acid assisted
condensation and simultanous water removal via azeotropic solvent
distillation, which is followed with DDQ quenching of the
porphyrinogen intermediate, but using liquid-liquid two phase
oxidation and crystallization for improved product isolation.We
obtained porphyrin 2a [900 mg (18.8%)] in pure state with mp = 248-249°C.
The success of this novel procedure may be related to coordination
complex formation between oxophilic La3+ cations and the precursor carbinols (template-effect). Without LaCl3 no detectable porhyrin formation was observed, but complete
decomposition of the carbinol when H2SO4*SiO2 was introduced.
Method_C:
Clay
supported acid [21]
catalyzed carbinol cyclization at high dilution in toluene with
simultanous water removal via azeotropic solvent distillation,
followed with DDQ quenching of the porphyrinogen intermediate, but
using liquid-liquid two phase oxidation and crystallization for
improved product isolation. Samples of pure(C7F15)4porH2 and (CF3)4porH2 with sharp meting
points are easily obtained at the gram scale in 15 or 25% yields,
respectively.
Experimental
Pyrryl-carbinols were prepared by NaBH4 reduction of the precursor ketones as shown below and they stored at 0°C until used. Solvents and reagents were purchased from Sigma-Aldrich and Molar. 1H-, 13C- and 19F-NMR spectra were recorded on Bruker Avance 250 instrument using a 5 mm inverse 1H/13C/31P/19F probe head at room temperature. Chemical shifts (δ) are given in parts per million (ppm) units relatively to the internal standard TMS (δ=0.00 for 1H, δ=0.00 for 13C) and to CFCl3 as external standard (δ=0.00 for 19F). Melting points were determined on a Boetius micro-melting point apparatus and are uncorrected. The reactions were monitored by TLC (SiO2 and Al2O3 plates), UV-Vis spectra (Carry) and gas chromatography (Hewlett-Packard 5890 Series II, PONA [crosslinked methylsilicone gum] 50 m x 0.2 mm x 0.5 μm column, H2 carrier gas, FID detection; Program: 120 °C, 5 min, 10 °C/min, 250 °C, 5 min; Inj.: 250 °C, Det.: 280 °C).
2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-Pentadecafluoro-1-(2’-pyrrolyl)-1-octanone (3a)
To a solution of 2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-pentadecafluorooctanoyl chloride (15.15 g, 0.035 mol) in ether (35 mL) was added dropwise the solution of pyrrole (2.41 g, 0.036 mol) in ether (30 mL) under stirring over 30 min period. After the addition the mixture was stirred for an additional 2 h at rt. The reaction mixture was washed with 5% NaHCO3 solution (2 x 50 mL) and water then dried over sodium sulfate. The solvent was removed then the crude product was crystallized from pentane to yield 8.2 g (57 %) pure product. Mp. 48-49 °C.
1H NMR (250 MHz CDCl3) δ (ppm): 6.43 (1H, m), 7.28 (2H, m), 9.98 (1H, s).
2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-Pentadecafluoro-1-(2’-pyrrolyl)-1-octanol (1a)
To an ice water cooled and stirred solution of 2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-pentadecafluoro-1-(2’-pyrrolyl)-1-octanone (9.27 g, 0.020 mol) in a solvent mixture (130 mL tetrahydrofurane and 60 mL methanol) was carefully added NaBH4 (1.76 g, 0.046 mol) in small portions during 30 min. After stirring for 2 h at rt ether (50 mL) was added and the solution was washed with saturated NaHCO3 (50 mL) then brine. The organic phase was dried over Na2SO4. After removal of the solvent the residue was crystallized from toluene (40 mL) to give 7.0 g (75%) pure crystalline product, mp = 93-95 °C.
1H NMR (250 MHz CDCl3) δ (ppm): 2.95 (1H,s), 5.00 (1H, m), 6.23 (1H, m), 6.31 (1H, s), 6.83 (1H, m) 8.59 (1H, s).
2-Trifluoroacetyl-pyrrole (3b)
To an ice cold solution of trifluoroacetic anhydride (46 mL, 0.325 mol) in ether (400 mL) was added dropwise the solution of pyrrole (19.0 g, 0.283 mol) in ether (55 mL) under stirring over 2.5 h period. After the addition the mixture was stirred at 0°C for an additional 4 h. The reaction mixture was washed with water then dried (Na2SO4). The solvent was removed then the residue steam distilled. The distillate was cooled to 0 °C and the crystalline solid formed filtered, washed with cold water and dried to yield 32.7 g (71 %) product. Mp. 38-40°C (reported value [17] mp 46-47 °C/ obtained by vacuum sublimation).
1H NMR (250 MHz CDCl3) δ (ppm): 6.43 (1H, t), 7.26 (1H, s), 7.31(1H, s), 10.18 (1H, s)
2,2,2-Trifluoro-1-(2’-pyrrolyl)-1-ethanol (1b)
To an ice water cooled and stirred solution of 2-trifluoroacetyl pyrrole (25 g, 0.153 mol) in methanol (100 mL) was carfully added NaBH4 (12.5 g, 0.33 mol) in small portions during 3 h. After stirring for 1 h saturated NaHCO3 (50 mL) was added and the mixture was extracted with ether (5 x 50 mL) then the combined organic phase was washed with saturated NaHCO3, brine, and then dried over Na2SO4. After removal of the solvent the residue was distilled in vacuo to afford 20.3 g (80.4%) colourless oil of bp. 112-120°C/18 mm Hg; which turns to a white solid at room temperature, m.p. = 45-47°C.
1H NMR (250 MHz CDCl3) δ (ppm): 2.95 (1H, s), 5.01 (1H, q), 6.22 (1H, m), 6.31 (1H, s), 6.83 (1H, m), 8.59 (1H,s).
Montmorillonite K10/TsOH*H2O reagent [Clay/TsOH (4:1 w/w)]
Montmorillonite K10 (50 g) was mixed with a solution of p-toluenesulfonic acid monohydrate (10 g) dissolved in distilled water (150 mL). The mixture was homegenized by evaporating most of the water using Rotavapor, then was further dried in vacuum over P2O5 for 3 days. The yield is quantitative (60 g).
H2SO4/SiO2 reagent [10% w/w H2SO4/SiO2]
Sulfuric acid (5.00 g, 96%) was mixed in a flask with SiO2 (45 g, Merck Kieselgel 40, 35-70 mesh). Some heat was evolved. The flask was stoppered and hand-shaked until a free-flowing reagent obtained. Then heated to 80°C and vacuum applied (16 mmHg) for 5 min. Finally allowed to cool to room temperature and stored in a well stoppered flask. Assay: ~1.0 mmol H2SO4/g solid.
meso-Tetrakis(perfluoroheptyl)porphin (2a)
Title compound was prepared by Method A using 5×10-3 Mcarbinol in the presence of 2.5×10-3 M TsOHcatalyst. Under an N2 atmosphere 3.40 g (7.31 mmol) C7F15-(α-pyrryl)-carbinol (1a); 1.5 L benzene and 0.72 g (3.78 mmol) TsOH*H2O were stirred and ½ vol of the solvent was distilled off. DDQ (1.73 g, 7.62 mmol) was added and refluxed 1 h. Filtered and evaporated to dryness. The residue was treated with CF2ClCFCl2 (700 mL) and HCO2H (350 mL) and 30% aq-H2O2 (35 mL) and refluxed for 5 h. Diluted with water (350 mL), phases were separated and the CF2ClCFCl2 layer was washed with 0.2 M NaOH (5 × 700 mL). Dried over Na2SO4 and the filtrate evaporated. The crude solid was recryst. from CF3C6H5/CH3OH to afford 230 mg (0.129 mmol, 7.1%) of 2a with m.p. = 248-249°C.
The spectroscopic data were in agreement with those reported in [11b]. 1H NMR (250 MHz, CDCl3, CF2ClCFCl2): δ 9.54 (s, 8 H), -2.25 (s 2 H); 19F NMR (CDCl3): δ-80.7, (br, 2 F), -82.3, (br, 3 F), -115.3, (br, 2 F), -121.7, (br, 2 F), -122.1, (br, 2 F), -122.8, (br, 2 F), -126.7, (br, 2 F); Vis(CF2ClCFCl2) of (Rf7)4PorH2: λmax = 404 nm (εmax= 143,760 Soret); [(Rf7)4porH4]2+: λmax = 423 nm (dication formation by CF3CO2H resulted in 19 nm red shift).
meso-Tetrakis(perfluoroheptyl)porphin (2a)
Title compound was prepared by Method B using 2×10-3 M carbinol and 1×10-3 M LaCl3*7H2O template in toluene.Under N2 atmosphere a mixture of 1a (5.00 g, 10.75 mmol), LaCl3*7H2O (2.00 g, 5.39 mmol) and toluene (5 L) was stirred and heated to reflux with the aid of an electric heating mantle and about 1 L toluene was distilled off (~30 min). Faint purple colour was developed. More toluene (1 L) was added along with of 10%H2SO4@SiO2 (2.50 g; = 250 mg H2SO4, 2.55 mmol) stirred and refluxed longer and toluene (500 mL) distilled off during the next 30 min to display darkened colour. Then DDQ (2.50 g, 11.0 mmol) was added and the mixture refluxed for 1 h. The mixture was filtered while hot and evaporated (Rotavapor). This solid was washed with CH3OH (200 mL) and dried to yield 3.10 g of crude product. It was dissolved in CF2ClCFCl2(500 mL) and strirred with a mixture of HCO2H (250 mL) and 30% aq-H2O2 (25 mL) for 12 h. The separated lower phase was washed with 85% HCO2H (3×100 mL), then with water (3×200 mL), 1M KOH (3×200 mL) and finally with brine. Dried over Na2SO4, filtered and the solvent recovered by atmospheric distillation to afford an enriched solid porphyrin fraction: 1.76 g (~36%) after dried at 80°C/16 mm Hg. Two times recrystallization from a mixture of CF3C6H5 (12 mL) and CH3OH (5 mL) gave 900 mg (18.8 %) of analytically pure (Rf7)4PorH2 as dark purple needles; m.p. = 248-249°C. UV-Vis (R-113): Soret 404 nm (ε = 143,760).
meso-Tetrakis(perfluoroheptyl)porphin (2a)
Title
compound was prepared by Method C using 2×10-3 M carbinol and 5×10-4 M TsOH*momoK10 in toluene.
A mixture of toluene (5.0 L) and 1:5 w/w supported TsOH*H2O/Montmorillonite K10 (600
mg; 0.53 mmol TsOH) and of 1a (5.00 g, 10.75 mmol) was stirred and heated under a
slow stream of N2 and about 1 L of toluene distilled off during 1 h. Next the
mixture is treated with DDQ (3.50 g, 15.4 mmol) and boiled with stirring for 1h longer. The
reaction mixture was filtered on filter paper while hot. This toluene filtrate gave dark
precipitate at 20°C but significant amount of porphyrin remained in solution. It was evaporated
in vacuum (Rotavap) and the residue was leached with CH3OH (400 mL) to remove
any nonfluorous (quinone/hydroquinone) side products. The fluorous precipitates and residues
were combined by dissolving them in CF2ClCFCl2 (500 mL), then the CF2ClCFCl2 extract of the crude fluorous porphyrin and oligomeric pyrromethenes were stirred with a
mixture of HCO2H (250 mL) and 30% aq-H2O2 (25 mL) for 24
h. The two liquid phase system was worked up as follows: the separated CF2ClCFCl2 layer was washed consecutively with 85% HCO2H (100 mL), then H2O (2×100
mL), 1M KOH (3×200 mL) and brine (2×50 mL). The solution was dried (Na2SO4),
filtered and evaporated to give ~2.5 g enriched product. Repeated (2×) recrystallization
of that from a 3:2 v/vmixture of CF3C6H5 and CH3OH gave dark purple needles, 1.208 g (0.6778 mmol, 25.2%) of (Rf7)4PorH2,
with m.p. = 246-248°C.UV-Vis (R-113): Soret 404 nm (ε = 143,760).
meso-Tetrakis(trifluoromethyl)porphin (2b)
Title
compound was prepared by Method C using 6×10-3 M CF3-carbinol
and 3×10-4 M TsOH*momoK10 in toluene.
A mixture of toluene (5.0 L) and 1:5 w/w supported TsOH*H2O/Montmorillonite K10 (1.60
g; 1.40 mmol TsOH) and of 1b (5.00 g, 30.3 mmol) was stirred and heated under a slow
stream of N2 and about 1 L of toluene distilled off during 1 h. DDQ (6.0 g, 26.4
mmol) was added to the mixture and boiled for 1h. Then basic (Brockman I) Al2O3 (250 g) was added and the mixture filtered while hot and the filtrate was evaporated in vacuum
(Rotavap). The residue was dissolved in CF2ClCFCl2 (500 mL) and then
stirred with a solution of 30% H2O2 (50 mL) in 98% HCO2H
(500 mL) at RT for 24 h. Liquid phases were separated, then the CF2ClCFCl2 layer was washed with H2O (2×100 mL), 1M KOH (3×200 mL) and brine (100 mL). The
CF2ClCFCl2 solution was dried (Na2SO4), filtered
and evaporated to give ~2 g enriched product. It was recrystallised from CF3C6H5-CH3OH
(9:1 v/v), and then 2-propanol (25 mL/100 mg) to yield 670 mg (1.15mM, 15.2%) analytically
pure dark blue lustrous crystals; mp = 192-196°C.
The spectroscopic data were in agreement with those reported in [11b]. 1H NMR (250 MHz, CDCl3): δ 9.60 (s, 8H), -2.08 (s, 2H); 19F NMR (CDCl3): δ -38.41 (s, 3F). Vis (CH2Cl2): Soret 403 nm (ε = 120,226), 510, 545, 593, 649.
Acknowledgement
This study was supported by the Hungarian Scientific Research Foundation (OTKA K 062191 “Sustainable fluorous chemistry”)
References
- Enikolopyan, N. S.; Bogdanova, K. A.; Askarov, K. A.;Metal Complexes of Porphine and Azaporphine Compounds as Catalysts of Reactions Involving Oxidation by Molecular Oxygen; Russian Chemical Reviews, 1983, 52, 13-26; Translated from Uspekhi Khimii, 1983, 52, 20-42.
- Mansuy, D., Biomimetic catalysts for selective oxidation in organic chemistry, Pure & Appl. Chem., 1990,62, 741-746.
- (a) Li,G.;Chen, Y.; Missert, J. R.; Rungta, A.; Dougherty, T. J.;Grossman, Z. D.; Pandey, R. K.;Application of Ruppert’s reagent in preparing novel perfluorinated porphyrins, chlorins and bacteriochlorins, J. Chem. Soc.,Perkin Trans. 1, 1999, 1785–1787. (b) Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey R. K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy, Chem. Soc. Rev. 2011, 40, 340-362.
- (a) Li, L.-L.; Diau, E. W.-G. Porphyrin-sensitized solar cells, Chem. Soc. Rev. 2013, 42, 291-304; (b) Urbani, M.; Grätzel, M.; Nazeeruddin, M. K.; Torres, T. Meso-Substituted Porphyrins for Dye-Sensitized Solar Cells, Chem. Rev. 2014, 114, 12330-12396.
- Briza, T.; Kaplánek, R.; M; Havlik, M.; Dolensky, B.; Kejik, Z.; Martasek, P.; Král, V., Synthesis of Highly Functionalized Fluorinated Porphyrins; Supramolecular Chemistry, 2008, 20 (3), 237–242.
- (a) Král, V.; Králová, J.; Kaplánek, R.; Bríza, T.; Martásek, P., Quo vadis porphyrin chemistry?Physiol. Res. 2006, 55 (Suppl. 2): S3-S26. (b) Chen, L. D.; Zou, X. U.; Buhlmann, P. Anal Chem. 2012, 84, 9192-9198.
- (a) Belyaeva, E. V.; Sigan, A. L.; Druzhinina, I. E.; Ikonnikov, N. S.; Chkanikov, N. D.; A method of introducing fluorinated substituents in porphyrin structure by nucleophilic substitution of fluorine in meso-tetrakis(pentafluorophenyl)porphyrin and pentafluorobenzaldehyde with polyfluoroaliphatic alcohols. Fluorine notes, Vol. 5(102) 2015; (b)Aggarwal, A.; Singh, S.; Samson, J.; Drain, C. M. Adaptive Organic Nanoparticles of a Teflon-Coated Iron (III) Porphyrin Catalytically Activate Dioxygen for Cyclohexene Oxidation. Macromol. Rapid Commun. 2012, 33, 1220−1226.
- (a) Moore, K. T.;Horváth, I. T.; Therien, M. J. High-Pressure NMR Studies of (Porphinato) iron-Catalyzed Isobutane Oxidation Utilizing Dioxygen as the Stoichiometric Oxidant; J. Am. Chem. Soc.,1997,119 , 1791–1792; (b) Moore, K. T.; Horváth, I. T.;Therien, M. J., Mechanistic studies of (porphinato) iron-catalyzed isobutane oxidation. Comparative studies of three classes of electron-deficient porphyrin catalysts.Inorganic Chemistry, 2000,39 (15), 3125-3139.
- (a) Horváth, I. T.,Rábai, J. Facile Catalyst Separation without Water: Fluorous Biphase Hydroformylation of Olefins. Science1994, 266, 72-75; (b)Fluorous Multiphase Catalyst or Reagent Systems for Environmentally Friendly Oxidation or Hydroformylation or Extraction Processes. Horvath, I. T.; Rabai, J. (Exxon Research and Engineering Co., USA). Eur. Pat. Appl. (1995), 11 pp. EP 633062 A1 19950111; (c) Metal-Fluorinated and Metal-Perfluorinated Complexes as Catalysts and Extractants for Multiphase Systems. Horváth, I. T.; Rábai, J. (Exxon Research and Engineering Co., USA). U.S. (1999), 8 pp., US 5981422 A 19991109.
- (a) Horváth, I.T. A personal view of the history of fluorous chemistry. Chapter 2; pp. 5-10. In: Gladysz, J. A.; Curran, D.P.; Horvath, I. T. (Eds): Handbook of Fluorous Chemistry, Wiley-VCH, 2004, Weinheim, Germany; (b) Fluorous Chemistry, Volume Editor: Istvan T. Horvath, Vol. 308, 2012; in: Topics in Current Chemistry, Springer-Verlag Berlin Heidelberg 2012.
- (a) DiMagno, S. D.; Williams, R. A.; Therien, M. J., Facile Synthesis of meso-Tetrakis(perfluoroalkyl)porphyrins: Spectroscopic Properties and X-ray Crystal Structure of Highly Electron-Deficient 5,10,15,20-Tetrakis (heptafluoropropyl)porphyrin.J. Org. Chem., 1994, 59, 6943-6948; (b) Goll, J. G.; Moore, K. T.; Ghosh, A.; Therien, M. J., Synthesis, Structure, Electronic Spectroscopy, Photophysics, Electrochemistry, and X-ray Photoelectron Spectroscopy of Highly-Electron-Deficient [5,10,15,20-Tetrakis (perfluoroalkyl)porphinato]zinc(II) Complexes and Their Free Base Derivatives. J. Am. Chem. Soc., 1996, 118, 8344-8354.
- For an overview of synthetic startegies and methods for porphyrins, see: (a) Wijesekera, T. P.; Dolphin, D. ’Synthetic Aspects of Porphyrin and Metalloporphyrin Chemistry’ In: Roger A. Sheldon (Editor), Metalloporphyrins in Catalytic Oxidations, Marcell Dekker, New York, 1994, Chapter 7, pp. 193-239.
- Adler, A. D.; Longo, F. R.; J. Finarelli, J.; Goldmacher, J.; Assour, J.; L. Korsakoff. L.; A simplified synthesis for meso-tetraphenylporphine. J. Org. Chern.1967,32, 476-476..
- Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M., Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987,52, 827-836.
- (a) Wijesekera, T. P.; 5-Perfluoroalkyldipyrromethanes and Porphyrins Derived Therefrom; Can. J. Chem.1996, 74, 1868-1871; (b) Wijesekera, T. P.; Lyons, J. E.; Ellis, P. E., Jr.; Bhinde, M. V., Pophyrins and Metal Complexes Thereof Having Haloalkyl Side Chains,US 5,608,054 (1977). (c) Wijesekera, T. P.; Lyons, J. E.; Ellis, P. E., Jr..Porphyrins And Their SynthesI From Dipyrromethanes And Aldehydes,US 5,760,217 (1998).
- Kuroda, Y.; Murase, H.; Suzuki, Y.; Ogoshi, H., A new route for meso-substituted porphyrin, Tetrahedron Lett. 1989,30, 2411-2412.
- Cooper, W. D.; Synthesis 2-Trifluoroacetylpyrole; J. Org. Chem., 1958, 23, 1382-1382.
- Sessler, J. L.; Mozaffari, A.; Johnson, M. R., 3,4-Diethylpyrrole and 2,3,7,8,12,13,17,18-Octaethylporphyrin, Org. Synth. 1992, 70, 68-78.
- А. С.Сейкин, О. И. Койфан, Б. Д. Березин, Уличшенный Метод Замещенных Тетрафенилпорфинов,Химия Гетероциклических Соединений,1986, No. 6, c. 798-801.
- (a) DiMagno, S. D.; Williams, R. A.; Therien, M. J., Facile Synthesis of meso-Tetrakis(perfluoro-Alkyl)-porphyrins: Spectroscopic Properties and X-ray Crystal Structure of Highly Electron-Deficient 5,10,15,20-Tetrakis (heptafluoropropyl)porphyrin.J. Org. Chem., 1994, 59, 6943-6948; (b) Therien, M. J., DiMagno, S. D.; Pyrrolic Compounds, US 5,817,830 (1998); (c) Therien, M. J., DiMagno, S. D.; Electron-deficient Porphyrins and Processes and Intermediates for Preparing the Same, US 5,856,515 (1999); (d) Moore, K. T.; Fletcher, J. T.; Therien, M. J.; Syntheses, NMR and EPR Spectroscopy, Electrochemical Properties, and Structural Studies of [5,10,15,20-Tetrakis(perfluoroalkyl) porphinato]iron(II) and -iron(III) Complexes. J. Am. Chem. Soc. 1999, 121, 5196-5209.
- Onaka, M.; Shinoda, T.; Izumi, Y., Nolen, E. Porphyrin Synthesis in Clay Nonospaces. Chemistry Letters, 1993, 117-120.
Recommended for publication by V. Kornilov
Fluorine Notes, 2016, 104, 3-4
