Received: November 2015
DOI 10.17677/fn20714807.2015.06.04
Fluorine Notes, 2015, 103, 1-2
FLUORINATED PHENYL(TRIMETHYL)SILANES
V. E. Boykoab, E.V. Igumnovab.
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Russia, 119991, GSP-1, Moscow, V-334, ul. Vavilova, 28
bZAO NPO PiM-INVEST, Russia, 119991 Moscow, ul. Vavilova, 28
e-mail: boykii@mail.ru
Abstract: This review summarisez the works related to the preparative methods and reactivity of fluorinated phenyl(trimethyl)silanes.
Keywords: fluorinated phenyl(trimethyl)silanes, polyfluoroaryl(trimethyl)silanes, pentafluorophenyl((trimethyl)silane.
The introduction of fluorine into a molecule influences on the physical, chemical, and biological properties of compounds. Due to this influence, organofluorine compounds are widely used in pharmaceutics, agrochemistry, and the producing various functional materials.
Great attention is paid to the development of synthesis methods and the studies of fluoroarylating reagents reactivity. Compounds that contain pentafluorophenyl moiety are used in medicinal chemistry, electron transport devices, light-emitting diodes, printing, liquid-crystal displays, and fluorine two-phase ligands [1-3].
Currently, pentafluorophenyl(trimethyl)silane and other polyfluorophenyl(trimethyl)silanes are considered prospective reagents for the introduction of pentafluorophenyl and polyfluorophenyl moieties into molecules of organic compounds [28-83], this is associated with their nontoxicity, stability and availability and with the fact that when they are used, substrates have significantly less limitations than in the cases of using traditional reagents, such as organolithium and organomagnesium compounds [4-7].
This review sums up the results of studying the syntheses of fluorinated aryl(trimethyl)silanes and their reactivity.
1. PREPARATION OF FLUORINATED PHENYL(TRIMETHYL)SILANES
1.1. Preparation of Fluorinated Phenyl(trimethyl)silanes Using Organometallic Derivatives
Pentafluorophenyl(trimethyl)silane was first mentioned in 1964. The group of Fild and co-workers showed that Grignard reagent prepared from bromopentafluorobenzene under mild conditions reacts with trimethylchlorosilane to give pentafluorophenyl(trimethyl)silane in 42% yield [4].
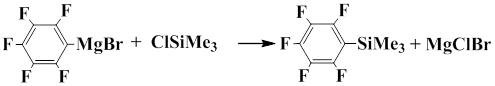
In 1969-70, Jukes described an analogous reaction with Grignard reagent obtained from chloropentafluorobenzene [8, 9].
In 1968, Respes and co-workers showed that bis(pentafluorophenyl)magnesium similarly reacts with trimethylchlorosilane to give pentafluorophenyl(trimethyl)silane in 86% yield [11]. The authors used organomagnesium compounds obtained by the exchange reaction of bromopentafluorobenzene with ethylmagnesium bromide and diethylmagnesium in tetrahydrofuran in 99% yield. It was shown that pentafluorobenzene also reacts with diethylmagnesium to form bis(pentafluorophenyl)magnesium [10].
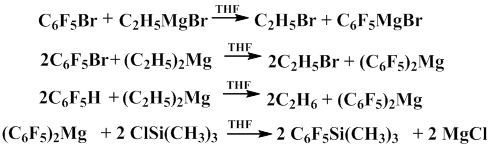
Perfluorophenylmagnesium compounds obtained from hexafluorobenzene and ethylmagnesium bromide in the presence of catalysts, such as CoCl2, FeCl2, NiCl2, CuI, TiCl4, PdCl2, RhCl3 and AgCl also react with trimethylchlorosilane in THF to give pentafluorophenyl(trimethyl)silane in 78% yield [10].
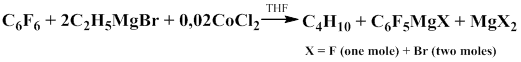
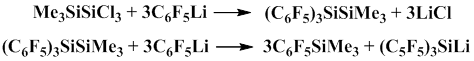
Lithium compounds can be also used to obtain pentafluorophenyl(trimethyl)silane. Weidenbruch and Dua described the methods for obtaining pentafluorophenyl(trimethyl)silane from pentafluorophenyllithium [12, 13].
Several independent groups discovered that fluorinated phenylsilanes can be obtained through the intermediate formation of fluorinated phenyllithium compounds from fluorinated benzenes. In this case, the end products are formed in 80% yield [14-17].
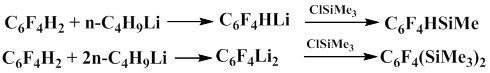
Miller and Sun showed that pentafluorophenyl silver, like Grignard reagent, reacts with trimethylchlorosilane to form pentafluorophenyl(trimethyl)silane.
![]()
Pentafluorophenyl silver was obtained via metal–metal or metal–halogen exchange reaction [18].
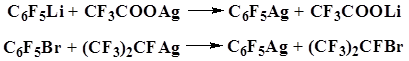
Seyferth and Hanson used pentafluorophenyl mercury derivatives as the starting compounds for the preparation of pentafluorophenyl(trimethyl)silane and obtained the desired product in 40% yield [19].
![]()
1.2. Preparation of Fluorinated Phenyl(trimethyl)silanes Using Fluoroaryl Halides and Fluorinated Benzenes
Bardin and co-workers studied the reactions of polyfluoroaromatic compounds with alkyl silicon halides in the presence of tris(dialkylamino)phosphine [20, 21]. The treatment of the mixture of bromopentafluorobenzene and trimethylchlorosilane with the equimolar amount of tris(diethylamino)phosphine at room temperature affords trimethylsilylpentafluorobenzene in 63-68% yield when pentane, or hexane, or dichlorometane is used as a solvent and in 30% yield when the reaction is carried out in benzene or acetonitrile [22].

Substitution of chlorine by Me3Si group in the polyfluoroaromatic ring under the action of P(NEt2)3 occurs much slower compared to the substitution of bromine anf iodine. When the substituent and the halogen are in a fixed position, the rate of silyldehalogenation is determined by the substituent nature.
It should be also noted that the absence of bis(trimethylsilyl)tetrafluorobenzenes in the products of the reaction with the equimolar ratio of reacting compounds C6F4X2 (X=Cl, Br, I) and trimethylchlorosilane points to high rate of silyldehalogenation of the latter, regardless of the halogen and its position with respect to Me3Si group. If the reaction is carried out with the excess of trimethylchlorosilane and tris(dialkylamino)phosphine, the product of substitution of two halogen atoms in the dibromotetrafluorobenzene molecule is obtained [20, 22].
Bader's group obtained bis(trimethylsilyl)tetrafluorobenzene in 49% yield when dihalogentetrafluorobenzene reacted with trimethylchlorosilane in the presence of magnesium in tetrahydrofuran and anhydrous trichloride iron is used as a catalyst [23].
![]()
In Weidenbruch’s works, it was shown that trimethyl(pentafluorophenyl)silane can be obtained by substitution of bromine in (tribromosilyl)pentafluorobenzene with methylmagnesium iodide when refluxed in diethyl ether for 48 hours. The product yield was 70% [24].
![]()
1.3. Preparation of Fluorinated Phenyl(trimethyl)silanes under UV Radiation and Using Electrochemical Methods
Kumar and co-workers described the preparation of bis(trimethylsilyl)tetrafluorobenzene in 48% yield from 1,2-bis(trimethylsilyl)ethyne and 3,4,5,6-tetrafluorocyclohex-3,5-dien-1,2-dione when exposed UV-radiation [25].
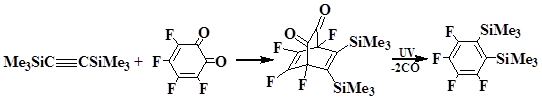
Stepanov and co-workers obtained pentafluorophenyl(trimethyl)silane by electrochemical silylation of fluoroaromatic compounds with trimethylsilane. Hexafluorobenzene, chloropentafluorobenzene and pentafluorobenzene served as the starting compounds, and reactions were carried out in acetonitrile and dimethylformamide with copper, nickel and steel cathode and zink and aluminium anode. The best result was achieved when a copper cathode and aluminium anode in acetonitrile were used. The yield of pentafluorophenyl(trimethyl)silane prepared from hexafluorobenzene was 60% [26].
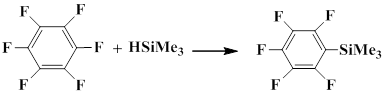
1.4. Preparation of Fluorinated Phenyl(trimethyl)silanes by the Decarboxylation of Fluoroaromatic Acid Derivatives
The method for preparation of polyfluoroaryl(trimethyl)silanes from commercially produced fluoroaromatic acids was developed under Igumnov’s guidance. When potassium salts of these acids react with trimethylchlorosilane in polar aprotic solvents, such as dimethylformamide, N-methylpyrrolidone or sulfolane, at 95–130°С, the corresponding polyfluoroaryl(trimethyl)silanes are formed in good yields.
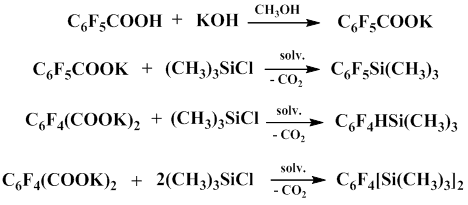
Тwo mechanisms for this process have been previously assumed. According to first mechanism, the salt of fluorinated aromatic acid is decarboxylated to give pentafluorophenyl anion, which later reacts with trimethylchlorosilane. The second mechanism includes the formation of trimethylsilyl ester of fluorinated aromatic acid as a result of the reaction between the salt of the corresponding acid with trimethylchlorosilane and the following decarboxylation of the resulting trimethylsilyl ester in the presence of a nucleophile, which is potassium chloride that is formed in the course of the reaction. The process involves the formation of pentacoordinate silicon intermediate.
Later it was discovered that trimethylsilyl esters of fluorinated aromatic acids are indeed decarboxylated in aprotic polar solvents in the presence of catalytic amounts of a nucleophile to give polyfluoroaryl(trimethyl)silanes.
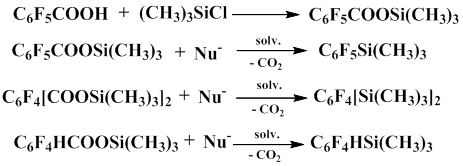
Thus, it was confirmed that the reaction proceeds through pentacoordinate silicon intermediate which, when heated, results in decarboxylation and the formation of polyfluoroaryl(trimethyl)silanes [27].
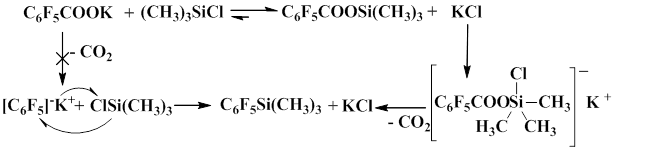
2. REACTIONS OF PENTAFLUOROPHENYL(TRIMETHYL)SILANE
2.1. Reactions with Carbonyl Compounds
Pentafluorophenyl(trimethyl)silane reacts with benzaldehyde in the absence of a catalyst at 165-170oC for 4 days to give silyl ester in 62% yield [28]. In the presence of the catalytical amount of KF, the reaction occurs under mild conditions for a shorter period of time.
Scientists from the Siberian Branch of the U.S.S.R. Academy of Sciences studied the reactions of pentafluorophenyl(trimethyl)silan with carbonyl compounds in the presence of cyanide-anion, specifically KCN complex with 18 crown-6-ether [29–42]. The authors studied the influence of carbonyl group, solvent, steric and electronic factors, and some other parameters on the course of the arylation process. They also explored the reaction mechanism involving aryl anion. It was also shown that when the reaction of pentafluorophenyl(trimethyl)silane with enolyzable carbonyl compounds is induced by cyanide-anion in diethyl ether, it gives trimethylsilyl ethers of the corresponding enols and pentafluorobenzene.
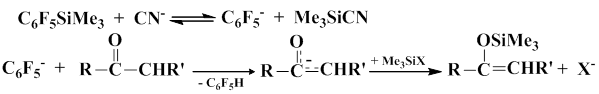
No. |
1 |
2 |
3 |
4 |
R |
tert-Bu |
Ph |
|
HC≡C |
R’ |
H |
H |
Et |
When this reaction is carried out in THF, it gives a polymeric product of molecular formula –(C6F4)n–.
![]()
Pentaflurorphenyl(trimethyl)silane reacts with carbonyl compounds not capable of enolyzing to give adducts whose hydrolysis leads to the formation of secondary alcohols.
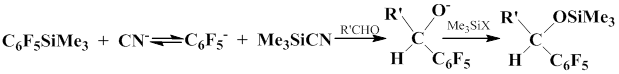
No. |
1 |
2 |
3 |
4 |
R’ |
Ph |
iso-Pr |
MeCH=CH |
PhCH=CH |
Yield, % |
56.5 |
37.3 |
56.4 |
40.1 |
Catalysis with Cu(dppe) complex also results in adducts of penfluorophenyl(trimethyl)silane to carbonyl group. In particular, the addition of C6F5SiMe3 to aldehydes followed by hydrolysis give the corresponding secondary alcohols according to scheme represented below.
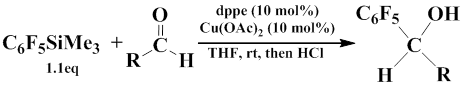
In this reaction, the following aldehydes such as 1-naphtaldehyde, 3-methylbenzaldehyde, isonitocinealdehyde, 2-nitrobenzaldehyde, thiophene-2-carbaldehyde, and 3-methoxybenzaldehyde were used [43].
Reactions of pentafluorophenyl(trimethyl)silane with carbonyl compounds are also catalyzed by fluoride-ion derived from anhydrous KF, CsF and [NBu4]F.
Such catalysis can be observed in the reactions of carboxylic acid chlorides with pentafluorophenyl(trimethyl)silane in the presence of either CsF or [NВu4]F, which afford arylketones.. These reactions can be used for preparation of carbinols.
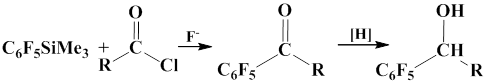
The addition of pentafluorophenyl group to benzaldehyde in the presence of tris(diethylamino)sulphonium salt of difluoro(trimethyl)silane (R2N)3S+ Me3SiF2- in THF was also reported. The authors state that the yield of the product is 87%, when this catalyst is used [44].
2.2. Reactions with Noncarbonyl Electrophiles
Fluoroaryl(trialkyl)silanes undergo desilylation when exposed to nucleophilic reagents, such as KF, CsF, and TBAF, this is used for selective introduction of functional groups into the aromatic ring. These reactions include protodesilylation, bromination, iodination, alkylation, and acylation.
CsF is the most effective catalyst in desilylation reactions according to the following scheme [45].

E |
Compounds obtained |
Yield, % |
I2 |
Iodopentafluorobenzene |
59 |
Br2 |
Bromopentafluorobenzene |
93 |
D2O |
Deuteropentafluorobenzene |
38 |
CH3I |
Pentafluorotoluene |
59 |
C6H5I |
Pentafluoro-4-methylbiphenyl |
50 |
C6H5HgOCOF3 |
Perfluorobiphenylmercury |
62 |
The authors also observed that other cesium and tetrabutylammonium salts does not catalyze protodesilylation reaction.
2.3. Preparation of Organometallic Aryl Derivatives
In 2002, German scientists scholars published the work where a method for the synthesis of pentafluorophenyl silver using pentafluorophenyl(trimethyl)silane as the source of the aryl group. The yield of the organometallic product was 97% [46].
![]()
This reaction was later used to obtain various bis(fluoroaryl)mercury derivatives [47].
![]()
No. |
1 |
2 |
3 |
4 |
5 |
Rf |
C6F5 |
2,3,4,6-F4C6H |
2,3,5,6-F4C6H |
2,4,6-F3C6H2 |
2,6-F2C6H3 |
Yield, % |
93 |
85 |
85.3 |
81.2 |
75.3 |
In addition, it was studied the possibility of formation cesium tetrakis(pentafluorophenyl)cesium indate in the reaction of pentafluorophenyl(trimethyl)silane with tris(pentafluorophenyl)indium complex in acetonitrile in the presence of the equimolar amount of cesium fluoride. The reaction proceeds through the intermediate formation of complex cesium salt of pentacoordinate silicon. This intermediate was further used to obtain [PNP][In(C6F5)4] complex [PNP=bis(triphenylphosphinanyliden)immonium] [48].
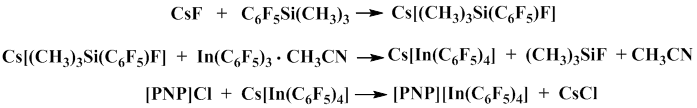
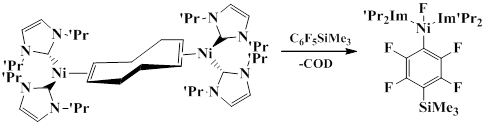
It was found that [Ni2(iPr2Im)4(COD)] substitutes the fluorine atom in the para-position of pentafluorophenyl ring. The reaction is carried out in THF at room temperature. trans-[Ni2(iPr2Im)2(F)(4-(SiMe3)C6F4)] was obtained in 61% yield [49].
2.4. Polymerization Reactions
Nishida's group developed the method of pentafluorophenylation of substituted pentafluorobenzenes with pentafluorophenyl(trimethyl)silane catalyzed with potassium bifluoride [50].
The same authors also showed that pentafluorophenyl(trimethyl)silane is convenient reagent for the pentafluorophenylation of 3,6-dihydro-3H-1,4-oxazine [51].
These reactions proceed via substitution of fluorine atom by pentafluorophenyl anion generated from pentacoordinate silicon intermediate formed as a result of interaction of pentafluorophenyl(trimethyl)silane with Lewis base.
2.5 Reactions of Silanes with Electrophilic Compounds Comprising Multiple Bonds
In the presence of cesium fluoride, pentafluorophenyl(trimethyl)silane reacts with terminal and internal perfluoroolefins with the substitution of one or two fluorine atoms in olefin to form perfluorinated phenylalkenes. The pentafluorophenylation of terminal perfluoroalkenes predominantly occurs at 20-22°С, whereas fluorine substitution in internal alkenes is observed only at 55-60°С [52].
No. |
Alkene |
Temperature, oC |
Time, h |
Conversion, % |
Product (yield, %) |
1 |
CF2=CF(CF2)4CFClCF2Cl |
20-22 |
24 |
51 |
C6F5CF=CF(CF2)4CFClCF2Cl (84) |
2 |
CF2=CFCF2C6F5 |
20-22 |
24 |
80 |
C6F5CF=CFCF2C6F5 (39) |
3 |
CF2=CFCF3 |
-10÷0 |
4 |
100 |
C6F5CF=CFCF3 (26); 4-C6F5C6F4CF=CFCF3 (45) |
4 |
C6F5CF=CFCF3 |
10 |
1 |
68 |
4-C6F5C6F4CF=CFCF3 (54) |
5 |
CF2=C(CF3)2 |
15 |
3 |
100 |
C6F5CF=C(CF3)2 (33); (C6F5)2C=C(CF3)2 (7); 4-C6F5C6F4CF=C(CF3)2 (10) |
6 |
(CF3)2C=CFC2F5 |
55-60 |
4 |
100 |
(CF3)2C=C(C6F5)C2F5 (63) |
The reaction with perfluorinated internal azaalkenes occurs in a similar manner [53].
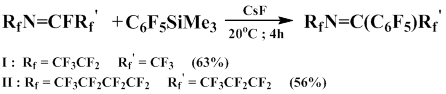
Starting azaalkene |
Product obtained |
Yield, % |
CF3(C2F5)NN=C(Cl)CF3 |
CF3(C2F5)NN=C(CF3)-C6F5 |
65 |
(CF3)2NN=C(Cl)C2F5 |
(CF3)2NN=C(CF3)C6F5 |
68 |
SF5N=C(Cl)C2F5 |
SF5N=C(C2F5)C6F5 |
55 |
Pentafluorophenyl(trimethyl)silane reacts with acetylenes to give trimethylsilylacetylenes and pentafluorobenzene [33].
![]()

Dilman and co-workers developed the method of nucleophilic pentafluorophenylation of arylidene malononitrile with pentafluorophenyl(trimethyl)silane. The reaction is carried out in DMF in the presence of sodium acetate as the main activator. The quantitative yield of the product was achieved when the reaction was carried out at room temperature for 3 h [54].
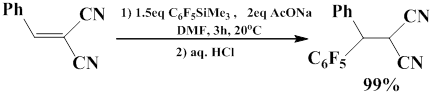
Another example of conjugated addition of fluorinated carbanion to the С=С bond is the reaction of 2-nitrocinnamate with pentafluorophenyl(trimethyl)silane. The reaction occurs in the presence of sodium acetate as Lewis base, which interacts with pentafluorophenyl(tromethyl)silane to generate the pentacoordinate intermediate, this further leads to the formation of pentafluorophenyl anion and its transfer to the electrophile with the formation of pentafluorophenylsubstituted 3-aryl-2-nitrobutanоates [55].
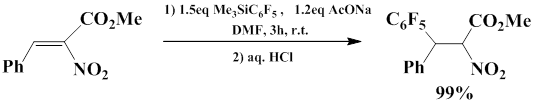
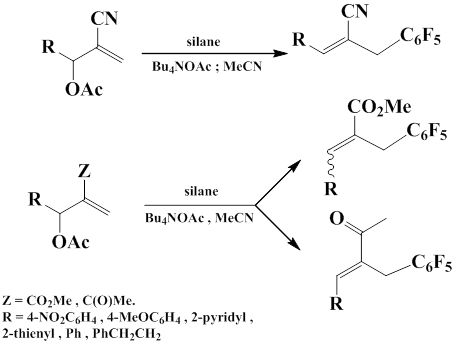
The studies were performed to explore how Baylis-Hillman reaction products containing nitrile, ester, or ketone groups react with C6F5-substituted silicon reagents (C6F5)4-nSiMen (n = 1–3). Reactions are induced by Bu4NOAc (5 mol%) in acetonitrile or in DMF under mild conditions and give the product of allyl substitution of acetoxy group by C6F5–carbanion in good yield. In all reaction one geometric isomer preferably or exclusively is formed (Z-isomer for nitriles and E-isomer for esters and ketones) [56].
Enamines react with pentafluorophenyl(trimethyl)silane in the presence of acids. The following mechanism was proposed for this process.
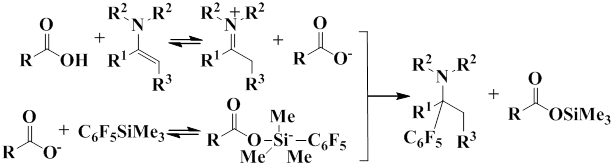
The scheme shows that at first the protonation of enamine with carboxylic acid occurs to form the iminium cation and the carboxylic acid anion. The cation interacts with pentafluorophenyl(trimethyl)silane to form a pentacoordinate particle, as a result the transfer of pentafluorophenyl anion to the iminium cation takes place.
The problem of realization of this mechanism consists in the the factors determining the concentration of electrophilic and nucleophilic particles, which are opposite. Stronger acids increase iminium cation concentration and simultaneously lower the nucleophilicity of carboxylic acid anion which is necessary for the formation of silicon intermediate. Moreover, protonation of this intermediate by acid can be a substantial side effect, which leads to the undesired formation of pentafluorobenzene.
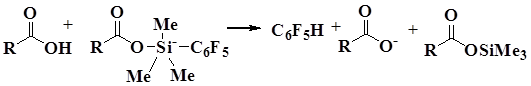
The reaction was carried out with enamines derived from pyrrolidone and piperidine which formed corresponding products in good yields (60-78%). However, in the case of substrates containing morpholine moiety, the yields of pentafluorophenylation products did not exceed 51% due to the electron-withdrawing effect of the oxygen atom, which destabilizes the iminium cation and lowers its equilibrium concentration [57].
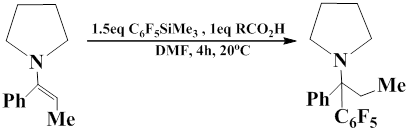
Pentafluorophenyl(trimethyl)silane reacts with iminium salts in DMF in the presence of Lewis bases, such as NaOAc or KF. The reaction leads to the formation of a new С – С bond and gives amines that contain a pentafluorophenyl group in α-position.
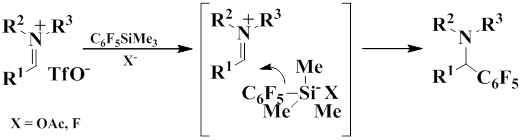
In turn, iminium salts were obtained by coupling from aldehydes, N-trimethylsilylamines, and TMSOTf,
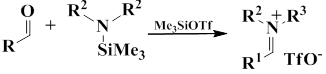
Iminium triflate salt was generated by this method from benzaldehyde and N-trimethylsilylpirrolidine in the presence of trimethylsilyl triphthalate in dichloromethane, pentafluorophenylation of this salt gave 1-((pentafluorophenyl)(phenyl)methyl) pirrolidone in 85% yield.
Another method for the synthesis of iminium salt is the methylation of imines with MeOTf [58].
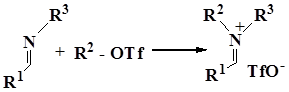
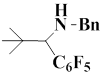
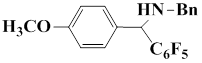
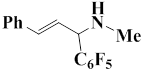
The following Table represents the examples of imines that enter the reaction and the yields of end products obtained from the reaction of iminium salt with pentafluorophenyl(trimethyl)silane:
No. |
Imine |
Product |
Yield, % |
1 |
|
|
80 |
2 |
|
|
85 |
3 |
|
|
94 |
One route for activation of the slightly reactive double bond C=N involves the formation of difluoroboron chelates by the reaction of boron trifluoride etherate with salicyl aldimines. In the presence of the catalytic amount of sodium acetate, pentafluorophenyl(trimethyl)silane nucleophilically pentafluorophenylizes the boron complex to give 2-[(methylamino)(pentalfluorophenyl)methyl]phenol [59].
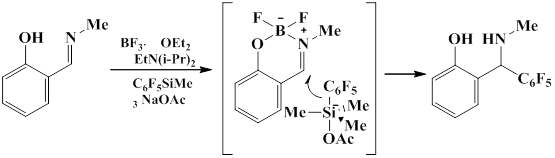
The method for pentafluoroarylation of tosyl imines was reported that uses phase carrier PhONa. The activation of silicon reagent was achieved by using the catalytic amount of TBAB as Lewis base [60].
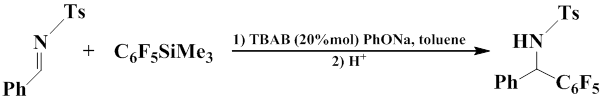
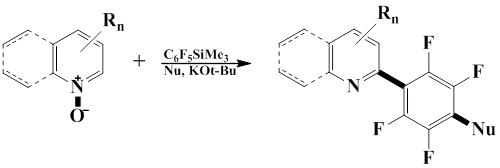
The the method, conditions, and synthetic characteristics of the reaction of heterocyclic N-oxides with pentafluorophenyl(trimethyl)silane were studied, which affords 2-perfluoroaryl substituted nitrogen-containing heterocycles in good yield [61].
2.6. Reactions with Sulfur Derivatives
Sulfimides readily react with pentafluorophenyl(trimethyl)silane under the same conditions as azaalkanes to form the product in sufficiently high yield (52%) [62].
![]()
Kirchmeier’s group showed that the reaction of compounds having S, O and active S-Cl or S-F bond with pentafluorophenyl(trimethyl)silane in the presence of KF or TBAF gives pentafluoroaryl sulfur derivatives, such as pentafluorophenylsulfoxides in 75-95% yield [63].

Like perfluoroalkylsilanes, pentafluorophenyl(trimethyl)silane readily reacts with SO3 under mild conditions to form pentafluorophenyl(trimethyl)silyl sulfonate in 69% yield [64].
![]()
Yagupolskii and co-workers showed that carbon sulfide reacts with pentafluorophenyl(trimethyl)silane in the presence of tetramethylammonium fluoride as a fluoride-ion source to afford tetramethylammonium pentafluorodithiobenzoate in 92% yield. The following alkylation of the salt with alkyl iodides gives corresponding ester of dithiocarboxylic acid in 35% yield [65].
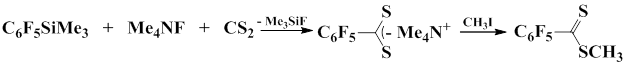
2.7 Reactions with XeF2
It has been established that pentafluorophenyl(trimethyl)silane does not react with a solution of xenon difluoride in acetonitrile at 20-40°С; however, the addition of the catalytic amount of cesium fluoride results in pentafluorobenzene decafluorobiphenyl, fluorosilane and xenon. Other polyfluoroaromatic alkyl silicon derivatives react similarly.
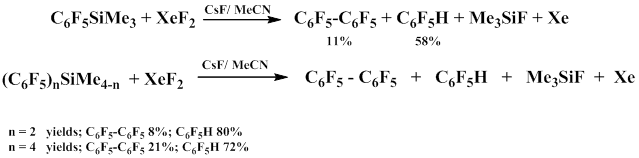
It was also found that in addition to usual products, in acetonitrile–benzene and acetonitrile–tribromomethane systems, 2,3,4,5,6-pentafluorodiphenyl and bromopentafluorobenzene are formed respectively.
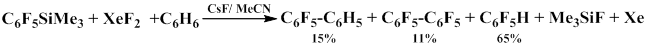
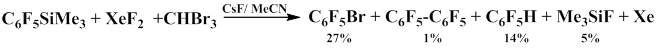
The products obtained indicate that the reaction proceeds via pentafluorophenyl radical formation [66, 67].
Naumann and Frohn group established that pentafluorophenyl(trimethyl)silane reacts with xenon fluoride in the presence of fluoride-ion, specifically [MeN4]F, to form bis(pentafluorophenyl)xenon. The reaction includes two steps. At the first step monosubstitution occurs to give pentafluorophenyl xenon fluoride that was recorded by NMR-spectra, then in the presence of the second equivalent of pentafluorophenyl(trimethyl)silane the second fluorine atom is substituted with the pentafluorophenyl group. If the second step uses RSiMe3-containing compounds where R ≠ C6F5, it results in the formation of covalent compounds of the C6F5XeR type where R ≠ C6F5, which are more stable than bis(pentafluorophenyl)xenon. Like its alkyl analogues, bis(pentafluorophenyl)xenon is not stable in organic solvents even at -45°С and decomposes to give decafluorobiphenyl and pentafluorobenzene [68-70].
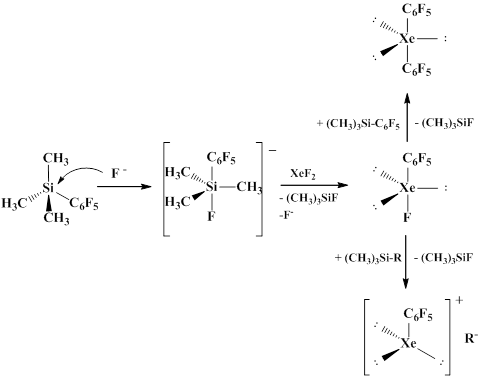
It was discovered that the reaction does not occur in the absence of a catalyst, which signifies that C6F5 в C6F5SiMe3 possesses insufficient nucleophilicity, and the basicity of fluorine atom in XeF2 is also insufficient for the formation of [C6F5SiMe3F]- anion. It was also shown that the type of products and their ratio are highly dependent on the concentration of starting reagents [71].
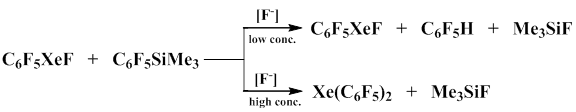
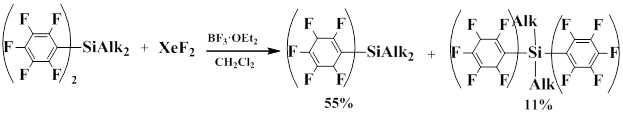
Bardin, Frohn and co-workers showed that compounds of formula (C6F5)nSiAlk4-n, where n=1,2 in the presence of BF3·OEt2 as fluoride-ion acceptor react with XeF2 to lead to polarization of the Xe-F bond, as a result 1-substituted 1,4-cyclohexadienes are formed.
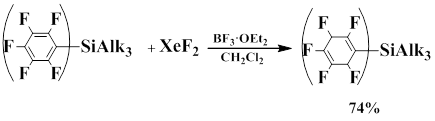
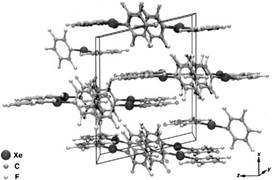
Then Neumann group studied and described the structure of bis(pentafluorophenyl)xenon and investigated the molecular packing in the crystal structure [72].
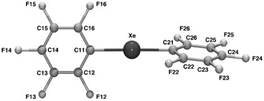
It was also shown that in the presence of the catalytic amount of tetramethylammonium fluoride, the substitution of fluorine in (2,6-difluorophenyl)fluoroxenon by penta- and tetrafluorophenyl group of the corresponding pentafluoro- and tetafluorophenylsilanes occurs to give diaryl xenon derivatives [73].
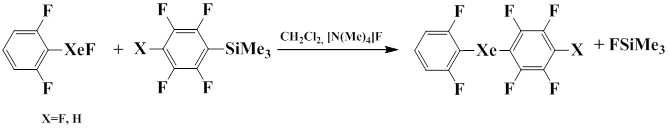
2.8 Reactions with Halogen Fluorides
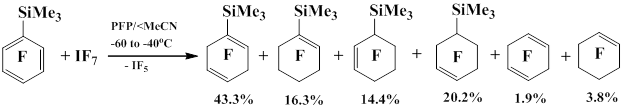
Pentafluorophenyl(trimethyl)silane reacts with BrF3 [74] and BrF5 [75] in CH2Cl2 in the presence of MeCN as a base to give C6F5BrFn (n = 2, 4). Under similar conditions, pentafluorophenyl(trimethyl)silane does not react with IF3 [76] и IF5 [77]. It was also discovered that the addition of pentafluorophenyl(trimethyl)silane, MeCN in a PFP solution to a solution of IF7 in PFP at -60°С to -40°С results in the slow addition of fluorine to the pentafluorophenyl derivative to afford corresponding heptafluorocyclohexa-1,4-dien-1-yltrimethylsilane, nonafluorocyclohexa-1-en-1-yltrimethylsilane, nonafluorocyclohexa-1-en-3-yltrimethylsilane, nonafluorocyclohexa-1-en-4-yltrimethylsilane, octafluorocyclohexa-1,4-diene and decafluorocyclohexa-1-ene [78].
It was found that C6F5SiMe3 reacts with BrF3 in CH2Cl2 at 0°С to give C6F5BrF2 in 58% yield. The studies indicate that in the presence of boron trifluoride etherate this reaction gives[BF4] as the major product, C6F5Br and Me3SiF are also formed in the reaction [79].
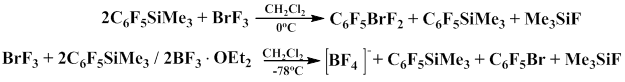
2.9. Reactions of Pentafluorophenyl(trimethyl)silane with Nucleophiles
The information concerning the reactions of nucleophilic reagents with fluoroaromatic silicon derivatives is very scarce. There are examples of pentafluorophenyl(trimethyl)silane reactions with potassium fluoride in ethanol that lead to pentafluorobenzene in literature [80]. Phenyl lithium reacts with pentafluorophenyl(trimethyl)silane to give product of substituting the fluorine atom in the para-position [81]. At the same time, when exposed to O- and S-nucleophiles, pentafluorophenyl(trimethyl)silane first undergoes protodesilylation, and then the resulting pentafluorobenzene interacts with nucleophile to give the products of substitution of the fluorine atom in the para-position. Pentafluorophenyl(trimethyl)silane reacts with piperidine used as N-nucleophile in two parallel ways depending on the temperature, namely substitution of the fluorine in the para-position and protodesilylation [82].

2.10. Oxydation of Pentafluorophenyl(trimethyl)silane Giving Pentafluorophephenol
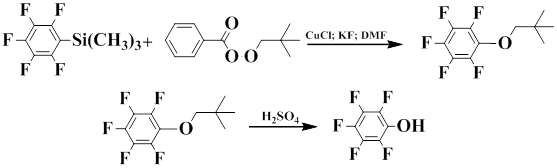
Igumnov and co-workers showed that pentafluorophenyl(trimethyl)silane is capable of oxidizing in the presence of tert-butyl peroxybenzoate to afford tert-butoxypentafluorobenzene, its hydrolysis gives pentafluorophenol in 65% yield [83].
References
- Montes V.A., Li G., Pohl R., Adv. Mater., 2004, 16, 2001-2003.
- Matsui M., Shirai K., Tanaka N., Funabiki K., Muramatsu H., Shibata K., Abe Y.,Ohgomori Y., J. Mater. Chem., 1999, 9, 2755-2763.
- Malhotra S.L., US рatent No 5744273 (1999).
- Fild M., Glemser O., Christoph G., Angew. Chem., 1964, 76, 953.
- Edmondson R.C., Jukes A.E., Gilman H., J. Organometal. Chem., 1970, 25, 273-276.
- Oliver A.J., Graham W.A.G., J. Organometal. Chem., 1969, 19, 17-27.
- Fearon F.W.G., Gilman H., J. Organometal. Chem., 1968, 13, 73-78.
- Jukes A.E., Gilman H., J. Organometal. Chem., 1969, 17(1), 145-148.
- JukesA.E., Hilman H., Edmonson, J. Organometal. Chem., 1970, 25, 273-276.
- Respes W.L., Tamborski C., J. Organometal. Chem., 1969, 18, 263-274.
- Respes W.L., Tamborski C., J. Organometal. Chem., 1969, 11, 619-622.
- Weidenbruch M., Abrotat G., John K., Chem. Ber., 1971,104, 2124-2133.
- Dua S.S., Gilman H., Indian J. Chem. B, 1979, 17, 562-565.
- Fearon F.W.G., Gilman H., J. Organometal.Chem., 1967, 10, 535-537.
- Tamborski C., Soloski E.J., J. Organometal. Chem, 1969, 17, 185-192.
- Fearon F.W.G., Gilman H., J.Organometal. Chem., 1968, 13, 73-80.
- Harper Jr., Soloski E.J., Tamborski C., J. Org. Chem., 1964, 29, 2385-2389.
- Miller W.T., SunK.K., J. Am. Chem. Soc. , 1970, 92 , 6985-6987.
- Seyferth D., Hanson E.M., Prokai B., Cross R.J., J. Organometal. Chem ., 1970, 24 , 33-43.
- Bardin V.V., Izv. Akad. Nauk, Ser. Khim ., 1997, No. 4, 813-817.
- Bardin V.V., Pressman L.S., Rogoza L.N., Furin .G., J. Fluorine Chem , 1991, 53 , 213-231.
- Bardin V.V., Pressman L.S., Rogoza L.N., Furin G.G., Zh. Obshch. Khim>., 1992, 62 (10), 2342-2349.
- Bader S.L., Kessler S.N., Wegner H.A., Synthesis , 2010, 16 , 2759-2762.
- Weidenbruch M., Wessal N.,Chem. Ber ., 1972, 105 , 173-187.
- Kumar V., Ramanathan S., Sang D., J. Org. Chem ., 2012, 77 , 966-970.
- Stepanov A.A., Martynov B.I., Tomilov A.P., Electrokhimiya, 2000, 36, 210-218.
- Boyko V.E., Tyutyunov A.A., Don V.L., Igoumnov S. M., Fluorine notes, 2013, Vol. 6(91).
- Webb A.F., Sethi D.S., Gilman H., J. Organometal. Chem., 1970, 21(2), 61-62.
- Vyazankin N.S., Vyazankina O.A., Gostevskii B.A., Titova I.A., Lopyrev V.A., Kalikhman I.D., Zh. Obshch. Khim., 1984, 54 (2), 461.
- Reutov O.A., Tetrahedron, 1978, 34(19), 2827-2855.
- Gostevskii B.A., Kruglaya O.A., Vyazankin N.S., Izv. Akad. Nauk SSSR, Ser. Khim., 1978, No. 10, 2425.
- Kruglaya O.A., Gostevskii B.A., Kalikhman I.D., Vyazankin N.S., Zh. Obshch. Khim.,1979, 49 (2), 354-360.
- Gostevskii B.A., Kruglaya O.A., Albanov A.I., Vyazankin N.S., J. Organometal. Chem., 1980, 187, 157-166.
- Gostevskii B.A., Vyazankina O.A., Kalikhman I.D., Bannikova O.B., Vyazankin N.S., Zh. Obshch. Khim.,1983, 53 (1), 229-230.
- KalikhmanI. D., Gostevskii B.A., Bannikova O.B., Vyazankin N.S., Vyazankina O.A., Izv. Akad. Nauk SSSR, Ser. Khim.,1983, No. 7, 1515-1518.
- Gostevskii B.A., Vyazankina O.A., Kalikhman I.D., Bannikova O.B., Vyazankin N.S., Zh. Obshch. Khim.,1983, 53 (9), 2051-2054.
- Gostevskii B.A., Vyazankina O.A., Vyazankin N.S., Zh. Obshch. Khim., 1984, 54 (5), 1209-1210.
- Gostevskii B.A., Vyazankina O.A., Vyazankin N.S., Zh. Obshch. Khim., 1984, 54(11), 2613-2617.
- Gostevskii B.A., KalikhmanI. D., Vyazankina O.A., Vyazankin N.S., Bannikova O.B., Zh. Obshch. Khim.,1984, 54 (6), 1428-1429.
- Vyazankina O.A., Gostevsky B.A., Vyazankin N.S., J. Organometal. Chem., 1985, 292(1/2), 145-149.
- Vyazankina O.A., Vyazankin N.S., Gostevskii B.A., Izv. Akad. Nauk SSSR, Ser. Khim., 1985, No. 11, 2585-2589.
- Vyazankina O.A., Vyazankin N.S., Gostevskii B.A., Titova I.A., Lopyrev V.A., Kalikhman I.D., Zh. Obshch. Khim.,1984, 54 (2), 461.
- Brogan S., Carter N.B., Lam H.W., Synlett, 2010, No. 4, 615-617.
- Kremlev M.M., Tyrra W., Naumann D., Yagupolskii Y., J. Fluorine Chem., 2005, 126(9/10), 1327-1331.
- Bardin V.V., Stennikova I.V., Furin G.G., Zh. Obshch. Khim., 1983, 53 (4), 812-815.
- Tyrra W., Wickleder M.S., Z. Anorg. Allg. Chem., 2002, 628, 1841-1847.
- Naumann D., Schultz F., Z. Anorg. Allg. Chem., 2005, 631, 122-125.
- Choi Z.-H., Tyrra W., Adam A., Z. Anorg. Allg. Chem., 1999, 625, 1287-1292.
- Schaub T., Fisher P., Steffen A., Braun T., Radius U., Mix A., J. Am. Chem. Soc., 2008, 130, 9304-9317.
- Nishida M., Fukaya H., Hayakawa Y., Ono T., J. Fluorine Chem., 2010, 131, 1314–1321.
- Nishida M., Hayakawa Y., Ono T., Fujii K., Uekusa H., Helvetica Chimica Acta, 2006, 89, 2671–2685.
- Bardin V.V., Petrov V.A., Stennikova I.V., Furin G.G., Zh. Org. Khim., 1989, 25(1), 52-55.
- Petrov V.A., Bardin V.V., Grinevskaya V.K., Mysov E.I., Furin G.G., German L.S., Izv. Akad. Nauk SSSR, Ser. Khim., 1990, No. 4, 923-928.
- Dilman A.D., Levin V.V., Belyakov P.A., Struchkova M.I., Tartakovsky V.A., Tetrahedron Letters, 2008, 49, 4352-4354.
- Zemtsov A.A., Levin V.V., Dilman A.D., Struchkova M.I., Tartakovsky V.A., J. Fluorine Chem., 2011, 132, 378-381.
- Zemtsov A.A., Levin V.V., Dilman A.D., Struchkova M.I., Belyakov P.A., Tartakovsky V.A., Hu J., Eur. J. Org. Chem., 2010, 6779-6785.
- Dilman A.D., Gorokhov V.V., Belyakov P.A., Struchkova M.I., Tartakovsky V.A., Izv. Akad. Nauk, Ser. Khim., 2007, No. 8, 1466-1468.
- Levin V.V., Kozlov M.A., SongY.-S., Dilman A.D., Belyakov P.A., Struchkova M.I., Tartakovsky V.A., Tetrahedron Letters, 2008, 49, 3108-3111.
- Dilman A.D., Arkhipov D.E., Levin V.V., Belyakov P.A., Korlyukov A.A., Struchkova M.I., Tartakovsky V.A., J. Org. Chem., 2007, 72, 8604-8607.
- Bernardi L., Indrigo E., Pollicino S., Ricci A., Chem. Commun., 2012, 48(10), 1428-1430.
- Stephens D. E., Chanez G., Valdes M., Dovalina M., Arman H.D., Larionov O.V., Org. Biol. Chem., 2014, 12, 6190-6199.
- Patel N.R., Kirchmeier R.L., Shreeve J.M., Inorg. Chem., 1993, 32 (22), 4802-4806.
- Patel N.R., Kirchmeier R.L., Inorg. Chem., 1992, 31(12), 2537-2540.
- Holfter H., Kirchmeier R.L., Shreeve J.M., Inorg. Chem., 1994, 33, 6369-6372.
- Babadzhanova L.A., Kirij N.V., Yagupolskii Y.L., J. Fluorine Chem., 2004, 125, 1095-1098.
- Bardin V.V., Stennikova I.V., Furin G.G., Leshina T.V., Yakobson G.G., Zh. Obshch. Khim.,1988, 58 (2), 2580-2588.
- Bardin V.V., Stennikova I.V., Furin G.G., Leshina T.V., Yakobson G.G., Zh. Org. Khim.,1985, 21 (2), 458-459.
- Maggiarosa N., Naumann D., Tyrra W., Angew. Chem. Int. Ed., 2000, 39 (24), 4588-4591.
- Frohn H.-J., Theissen M., Angew. Chem. Int. Ed., 2000, 39 (24), 4591-4593.
- Frohn H.-J., Organometallics, 2001, 20, 4750-4762.
- FrohnH.-J., Theissen M., J. Fluorine Chem.,2004, 125(6), 981-988.
- Bock H., Hinz-Hubner D., Ruschewitz U., Naumann D., Angew. Chem. Int. Ed., 2002, 41(3), 448-450.
- Bock H., Scherer H., Tyrra W., Naumann D., J. Fluorine Chem., 2006, 127, 1440-1445.
- Frohn H.-J., Schrinner K., Z. Anorg. Allg. Chem., 1997, 623 , 1847-1849.
- Breuer W., Frohn H.-J., J. Fluorine Chem>., 1990, 47 , 301-315.
- Frohn H.-J., Schrinner K., Z. Anorg. Allg Chem ., 1997, 623 , 1847-1849.
- Frohn H.-J., Chem. Ztg ., 1984, 108, 146-147.
- Frohn H.-J., BardinV.V., J. Fluorine Chem., 2010, 131, 1000-1006.
- Frohn H.-J., Bardin V.V., J. Fluorine Chem., 2010, 131(10), 922-932.
- Chambers R.D., Chivers T., J. Chem. Soc., 1964, No. 12, 4782-4790.
- Fearon F.W.G., Gilman H.J., J. Organometal. Chem., 1968, 13(1), 73-80.
- Bardin V.V., Stennikova I.V., Furin G.G., Zh. Obshch. Khim.,1987, 57 (7), 1580-1583.
- Boyko V.E., Tyutyunov A.A., Don V.L., Igoumnov S.M., Fluorine notes, 2013, Vol. 6(91).
Recommended for publication by Prof. S. M. Igumnov
Fluorine Notes, 2015, 103, 1-2
