Received: September, 2015
DOI 10.17677/fn20714807.2015.05.02
Fluorine Notes, 2015, 102, 3-4
A method of introducing fluorinated substituents in porphyrin structure by nucleophilic substitution of fluorine inmeso-tetrakis(pentafluorophenyl)porphyrin and pentafluorobenzaldehyde with polyfluoroaliphatic alcohols
aElizaveta V. Belyaeva*, aAndrey L. Sigan, bIana E. Druzhinina, aNikolai S. Ikonnikov, aNikolai D. Chkanikov
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences,
ul. Vavilova 28, V-334, GSP-1, 119991 Moscow, Russia
e-mail:faftor.belyaeva@mail.ru
bD.I. Mendeleyev University of Chemical Technology of Russia, Miusskaya sq. 9, 125047
Moscow, Russian Federation
Fax: +7 (499) 978-86-60
Abstract: Various methods for obtaining meso-tetrakis(pentafluorophenyl)porphyrins, additionally modified with polyfluoroalkoxy substituents are considered. One of the variants for macrocycle assembling includes synthesis and condensation of polyfluoroalkylbenzaldehydes obtained by substitution of para- and ortho-fluorines in the starting pentafluorobenzaldehyde by polyfluoroaliphatic alcohols. Direct substitution of fluorines in meso-tetrakis(pentafluorophenyl)porphyrin by polyfluoroaliphatic alcohols was carried out as an alternative.
Keywords: Fluorine-containing porphyrins, meso-tetrakis(pentafluorophenyl)porphyrin, polyfluoroaliphatic alcohols, pentafluorobenzaldehyde, nucleophilic substitution of fluorine.
Porphyrin compounds play the key role in a number of chemical and biochemical processes. The possibility of introducing substituents of various nature in a porphyrin cycle essentially extends the range of their potential applications in various fields of chemical technology as catalysts, components of photoactive materials and pharmaceuticals. Recently the considerable attention has been paid to the studies of fluorinated porphyrins in connection with the development FBS (Fluorous Biphasic System) technology [1,2], the works directed to design of photoelectroactive materials having hydrophobic characteristics [3,4] and expanding the range of effective photosensitizers potentially applicable in photodynamic therapy of cancer [5]. It was reported that solubility in perfluorocarbon media and hydrophobicity of porphyrins are provided by introducing not less than four perfluoroalkyl substituents having 3 to 8 carbons [1,2] whereas the presence of trifluoromethyl substituents markedly affect the redox properties [6].
Currently, the main methods for preparing fluorinated porphyrins are catalytic alkylation with perfluoroalkyliodides, or introducing fluorine-containing moieties by nucleophilic substitutions [7]. For the reason of extending sphere of fluorinated porphyrins applications, a search for new methods for their synthesis still remains topical. Our approach for solving this problem consists in either use of polyfluoroaliphatic alcohols in substitutions of fluorines in pentafluorobenzaldehyde under the action of base followed by condensation of the prepared compounds to give a macrocycle (Method А), or the similar substitution of the activated fluorines in aromatic rings of meso-tetrakis(pentafluorophenyl) porphyrin (Method B).
Aldehyde-pyrrole condensation of polyfluoroalkoxy substituted benzaldehyde (Method A)
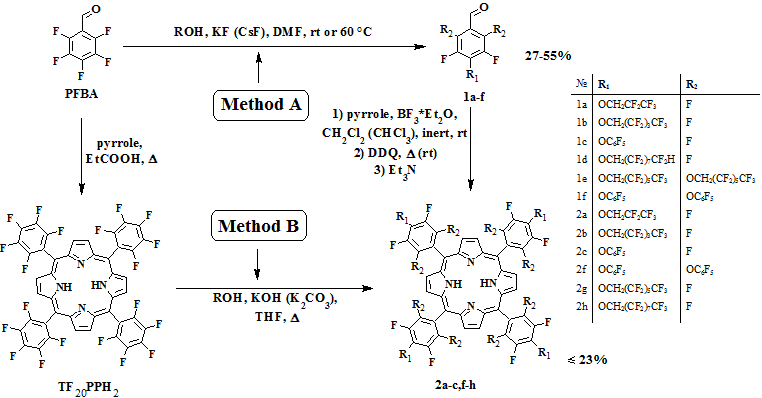
The substitution of the activated fluorine in para-position of pentafluorobenzaldehyde (PFBA) was earlier demonstrated by the example of a number of phenols [8], wherein such specific bases as potassium and cesium fluorides were used for formation of corresponding phenolates. We took this procedure as a base one for introducing polyfluorinated alcohol residues with relatively high acidity (рКа ~12-13) into PFBA molecule. Actually it was shown that interaction of PFBA with equimolar amounts of various polyfluoroaromatic alcohols or pentafluorophenol led to formation polyfluoroalkoxy- and phenoxy-benzaldehyde (1a-d). The reactions were carried out in dimethylformamide at room temperature up to complete conversion of the starting aldehyde (according to TLC data). It should be noted that in the case of three-fold excess of the corresponding alcohols, we didn’t observe substitution the ortho-fluorines in PFBA irrespective of the reaction time. At the same time the possibility of formation of the corresponding 2,4,6-substituted benzaldehyde (1e,f) in good yields was demonstrated in the case of increasing the reaction temperature to 60°С.
Aldehyde-pyrrole condensation of the prepared polyfluorophenoxy- and alkoxy-benzaldehydes (1a-c) was carried out using Lindsey method [9] recommended for the synthesis of porphyrins from aldehydes having bulk substituents. The reactions were carried in dichloromethane at room temperature in the presence of boron trifluoride diethyl etherate and using DDQ as an oxidizing agent. The corresponding symmetrical porphyrins (2a-c) were synthesized and characterized by NMR and mass spectroscopy. The yields were good for the compounds of this class.
According to literature data, the presence of even small substituents (for example methyl or methoxy) in positions 2 and 6 of benzaldehydes significantly hinders formation of porphyrins. Condensation of ortho-substituted benzaldehydes with pyrrole resulting to porphyrins requires the use of a cocatalyst (ethanol), as had been demonstrated by the example of mesitylaldehyde [10]. Application of this procedure to condensation of aldehyde (1f) with pyrrole led to formation of the corresponding porphyrin (2f), but the yield of this product did not exceed 2%. The attempt to carry out the reaction with aldehyde (1e) has failed, this can be explained either by the greater steric bulk of the substituents (1,1-Н,Н-perfluoroheptyloxy- in comparison with pentafluorophenoxy-group) or low solubility of intermediate compounds in chloroform that we observed in both cases.
Substitution of activated fluorines in aromatic rings of meso-tetrakis(pentafluorophenyl)porphyrin by polyfluoroalkoxy moieties (Method B) [11].
As we mentioned above, the reaction of meso-tetrakis(pentafluorophenyl)porphyrin (TF20PPH2) with various fluorine-containing N-, S-, and O-nucleophiles under the action of bases [7] can be also used as a method for introducing polyfluoroalkyl substituents in a porphyrin molecule. At the same time any examples with using of long-chain polyfluoroaliphatic alcohols in these reactions were not reported in literature, therefore we based on the procedure proposed for their non-fluorinated analogues [12].
The substitution reactions were carried out by heating TF20PPH2 in tetrahydrofuran with polyfluoroaliphatic alcohols in the presence of potassium hydroxide or potassium carbonate (see Scheme, Method B). The best results were achieved when HOCH2C6F13and HOCH2C8F17 were used, tetrasubstituted porphyrins (2g,h) were isolated in 37% and 47% yields, respectively. The formation of mono-, di- and tri-substituted porphyrins as the by-products of this reaction was observed (according to NMR and mass-spectroscopy), these products make it difficult to isolate and purify of the tetrasubstituted porphyrins. The use of substantial excess of the polyfluoroaliphatic alcohol in this reaction (by the example of HOCH2(CF2)2H) and increasing the reaction time didn’t result in selective formation of tetrasubstituted porphyrin. However in this case we observed that in the reaction mixtures contained the products of substituting fluorines not only in para-positions, but also in ortho- and meta-positions of pentafluorophenyl moieties.
Experimental
NMR spectra 1H and 19F were recorded on a Bruker AMX-400 and AMX-300 spectrometer with frequency 400.13 and 376.50 MHz at 20°C. Chemical shifts of 1H and 19F were determined relatively to proton signal of solvent (CDCl3) and trifluoroacetic acid (TFA) as external standard.
The ESI (1) and APCI (2) mass-spectra were registered on the Finnigan LCQ Advantage tandem dynamic mass-spectrometer. Nitrogen 10/0 (1) or 70/10 (2) served as a sheath and auxiliary gas. Flow rate acetonitrile 50 µl/min (1) or 350 µl/min (2).The temperature of the heated capillary was 150 °С, the electric potential between the needle and the counter electrode was 4.5 kV (1) or 6.0 kV (2). The samples with the concentration of 10-4 mol/l in acetonitrile solution were introduced into the ion source through the Reodyne injector with the 5 µl loop. Mass-spectra (EI-DIP) were recorded on mass-spectrometer Finnigan Polaris Q, ionization energy – 70 eV, procedure of sample injection – DIP.
Monitoring of the reaction course and purity of products were controlled by Merck Kieselgel 60 F254 plates. For column chromatography silica gel (MN Kieselgel 60) and aluminium oxide (neutral, 100-200 μm) were used. Elemental analysis was performed in the laboratory of elemental analysis INEOS RAS.
Reagents and solvents: tetrahydrofuran (THF), triethylamine (Et3N) and dimethylformamide (DMF) were dried with help of standard procedures. Chloroform and dichloromethane were purified by distillation over CaCl2 and CaH2 respectively. Pyrrole and boron trifluoride etherat were distilled before using. Potassium and cesium fluorides were annealed and stored in a sealed container. Polyfluorinated alcohols and pentafluorobenzaldehyde were purchased from “P&M Invest”. Meso-tetrakis(pentafluorophenyl)porphyrine was synthesized from pyrrole and pentafluorobenzaldehyde according to the literature procedure [13].
4-(1,1-H,H-perfluoropropyl-1-oxy)-2,3,5,6-tetrafluorobenzaldehyde (1a)
To a solution of 1,1-H,H-perfluoropropan-1-ol (3.37 g, 23 mmol) in DMF (40 ml) pentafluorobenzaldehyde (2.65 g, 20 mmol) and KF (2.65 g, 50 mmol) were added. Reaction mixture was stirred at room temperature for 5 h (reaction course was controlled by TLC). Then diethyl ether (70 ml) was added, and mixture was washed with saturated aqueous solution of NH4Cl (2 x 40 ml) and water (2 x 40 ml). Organic layer was separated and dried over CaCl2. After removing of solvent partially crystallizing yellow viscous liquid was obtained. Product was purified by flash-chromatography (aluminium oxide, hexane-chloroform = 1:1) to afford 2.77 g of light yellow liquid (yield 38%). 1H NMR (CDCl3, δ, ppm): 4.78 (t, 2H, J=12.08 Hz, OCH2), 10.20 (s, 1H, СНО). 19F NMR (CDCl3, δ, ppm): -78.85 (s, 2F, CAr-Forto), -68.04 (s, 2F, CAr-Fmeta), -47.35 (s, 2F, CF2), -6.34 (s, 3F, CF3). Anal. calcd for C10H3F9O2 (%): C, 36.83; H, 0.93; F: 52.43. Found (%): C, 36.48; H: 1.02; F, 52.12.
4-(1,1-H,H-perfluoropentyl-1-oxy)-2,3,5,6-tetrafluorobenzaldehyde (1b)
To a solution of 1,1-H,H-perfluoropentan-1-ol (4.21 g, 17 mmol) in DMF (40 ml) pentafluorobenzaldehyde (3.00 g, 15 mmol) and KF (2.05 g, 30 mmol) were added. Reaction mixture was stirred at room temperature for 5 h (reaction course was controlled by TLC). Then diethyl ether (70 ml) was added, and mixture was washed with saturated aqueous solution of NH4Cl (2 x 40 ml) and water (2 x 40 ml). Organic layer was separated and dried over CaCl2. After removing of solvent partially crystallizing yellow viscous liquid was obtained. Product was purified by flash-chromatography (aluminium oxide, hexane-chloroform = 1:1) to afford 1.97 g of light yellow liquid (yield 27%). 1H NMR (CDCl3, δ, ppm): 4.82 (t, 2H, J = 2.72 Hz, OCH2), 10.23 (s, 1H, СНО). 19F ЯМР (CDCl3, δ, ppm): -78.63 (d, 2F, J = 11.00 Hz, CAr-Forto), -67.79 (dd, 2F, J1 = 11.00 Hz, J2 = 8.25 Hz, CAr-Fmeta), -49.03 (s, 2F, CF2), -46.78 (s, 2F, CF2), -43.57 (s, 2F, CF2), -3.73 (s, 3F, CF3). MS (EI-DIP), m/z: 477.1 [M+H]+, 459.1 [C4F9CFCHOC6F4CHF]+, 193.2 [M-C5F11CH2]+, 151.2 [CF3CFHCF2]+.
4-(1,1,9-H,H,H-perfluorononyl-1-oxy)-2,3,5,6-tetrafluorobenzaldehyde (1d)
To a solution of 1,1,9-H,H,H-perfluorononan-1-ol (8.64 g, 20 mmol) in DMF (20 ml) pentafluorobenzaldehyde (3.92 g, 20 mmol) and KF (2.32 g, 40 mmol) were added. Reaction mixture was stirred at room temperature for 10 h (reaction course was controlled by TLC). Then diethyl ether (70 ml) was added, and mixture was washed with saturated aqueous solution of NH4Cl (2 x 40 ml) and water (2 x 40 ml). Organic layer was separated and dried over CaCl2. After removing of solvent partially crystallizing yellow viscous liquid was obtained. Product was purified by flash-chromatography (silica gel, ethyl acetate) to afford 6.72 g of light yellow solid (yield 55%). 1Н NMR (CDCl3, δ, ppm): 4.82 (t, 2H, J1 = 12.72 Hz, J2 = 12.47 Hz, OCH2), 6.08 (tt, 1H, J1 = 51.84 Hz, J2 = 5.14 Hz, CF2H), 10.27 (s, 1H, CHO). 19F NMR (CDCl3, δ, ppm): -78.03 (dd, 2F, J1 = 11.97 Hz, J2 = 8.98 Hz, CAr-Forto), -67.10 (dd, 2F, J =8.98 Hz, CAr-Fmeta), -59.34 (s, 2F, CF2), -51.63 (s, 2F, CF2), -45.64 (s, 2F, CF2), -45.44 (s, 2F, CF2), -44.29 (s, 6F, CF2CF2CF2), -42.92 (s, 2F, CF2). MS (EI-DIP), m/z: 608.3 [M]+, 193.2 [M-HCF2C7F14CH2]+.
4-(pentafluorophenyloxy)-2,3,5,6-tetrafluorobenzaldehyde (1c)
To a solution of pentafluorophenol (1.84 g, 10 mmol) in DMF (10 ml) pentafluorobenzaldehyde (1.96 g, 10 mmol) and CsF (2.84 g, 20 mmol) were added. Reaction mixture was stirred at room temperature for 2 h. Reaction mixture was poured into water and product was extracted by ethyl acetate. Organic layer was separated and dried over CaCl2. After removing of solvent partially crystallizing yellow viscous liquid was obtained. Product was crystallized from diethyl ether to afford 1.81 g of light yellow solid (yield 50%). 1Н NMR (CDCl3, δ, ppm): 10.28 (s, 1H, CHO). 19F NMR (CDCl3, δ, ppm): -83.26 (t, 2F, J = 20.53 Hz, C6F5O (meta-F)), -80.13 (t, 1F, J = 21.80 Hz, C6F5O (para-F)), -78.12 (d, 2F, J = 10.90 Hz, C6F4CHO (meta-F)), -77.89 (d, 2F, J = 19.62 Hz, C6F5O (orto-F)), -66.52 (dd, 2F, J1 = 10.90 Hz, J2 = 8.72 Hz, C6F4CHO (orto-F)). Anal. calcd. for C13HF9O2 (%): C, 43.36; H, 0.28. Found (%): C, 43.25; H, 0.28.
2,4,6-tri(1,1-H,H-perfluoroheptyl-1-oxy)-3,5-difluorobenzaldehyde (1e)
To a solution of 1,1-H,H-perfluoroheptan-1-ol (11.55 g, 33 mmol) in DMF (15 ml) pentafluorobenzaldehyde (1.96 g, 10 mmol) and KF (3.83 g, 66 mmol) were added. Reaction mixture was stirred at 60°С for 124 h (reaction course was controlled by TLC) and cooled down to room temperature. Then diethyl ether (50 ml) was added, and mixture was washed with saturated aqueous solution of NH4Cl (2 x 30 ml) and water (2 x 20 ml). Organic layer was separated and dried over CaCl2. After removing of solvent partially crystallizing yellow viscous liquid was obtained. Product was crystallized from dichloromethane to afford give 5.01 g of light-yellow solid (yield 42%). 1Н NMR (CDCl3, δ, ppm): 10.32 (s, 1H, CHO). 19F NMR (CDCl3, δ, ppm): -69.64 (s, 2F, CAr-Fmeta); -48.62 (s, 6F, CF2); -45.65 (s, 6F, CF2); -45.26 (s, 6F, CF2); -44.56 (s, 6F, CF2); -43.07 (s, 6F, CF2); -3.29 (m, 9F, CF3).
2,4,6-tri(pentafluorophenyloxy)-3,5-difluorobenzaldehyde (1f)
To a solution of pentafluorphenol (6.07 g, 33 mmol) in DMF (15 ml) pentafluorobenzaldehyde (1.96 g, 10 mmol) and KF (3.83 g, 66 mmol) were added. Reaction mixture was stirred at 60°С for 60 h (reaction course was controlled by TLC) and cooled down to rt. Then diethyl ether (50 ml) was added, and mixture was washed with saturated aqueous solution of NH4Cl (2 x 30 ml) and water (2 x 20 ml). Organic layer was separated and dried over CaCl2. After removing of solvent partially crystallizing yellow viscous liquid was obtained. Product was crystallized from dichloromethane to afford give 1.88 g of light yellow solid (yield 27%). 1Н NMR (CDCl3, δ, ppm): 10.48 (s, 1H, CHO). 19F NMR (CDCl3, δ, ppm): -83.06 (t, 4F, J1 = 21.36 Hz, J2 = 18.31 Hz, orto-OC6F5(para-F)); -79.64 (t, 1F, J 1= 21.36 Hz, J2 = 24.41 Hz, para-OC6F5(para-F)); -78.56 (d, 4F, J1 = 18.31 Hz, orto-OC6F5(orto-F)); -77.70 (d, 2F, J1 = 18.31 Hz, para-OC6F5(orto-F)); -69.53 (s, 2F, C6F2CHO(meta-F)). MS (EI-DIP), m/z: 520.0 [M-C6F5]+, 167.2 [C6F5]+.
meso-tetrakis(4-(2,2,3,3,3-pentafluoropropyl-1-oxy)-2,3,5,6-tetrafluorophenyl)porphyrine (2a)
A solution of 1a (1.2 g, 3.7 mmol) and pyrrole (0.25 g, 3.7 mmol) in dry dichloromethane (400 ml) was bubbled with argon for 30 min, BF3 x Et2O (0.14 ml, 1.1 mmol) was added dropwise, and reaction mixture was stirred at room temperature for 21 h. Then DDQ (0.92 g, 4.07 mmol) was added, and reaction mixture was refluxed for 2 h. After addition of Et3N (0.2 ml) solvent was removed, residue was washed with cold methanol to uncolored solution. Product was purified in 2 steps: 1) flash-chromatography (silica gel, dicloromethane), 2) column chromatography (silica gel, hexane-chloroform = 1:1) to afford 0.32 g of violet powder (yield 23%). 1Н NMR (CDCl3, δ, ppm): -2.89 (s, 2H, NH); 5.02 (t, 8H, J = 12.08 Hz), 8.96 (s, 8H, Pyr-H). 19F NMR (CDCl3, δ, ppm): -78,55 (m, 8F, CAr-Forto); -59.44 (m, 8F, CAr-Fmeta); -46.55 (s, 8F, CF2); -5,52 (s, 12F, CF3).MS (APCI), m/z: 1495.8 [M+H]+.
meso-tetrakis(4-(1,1-H,H-perfluoropentyl-1-oxy)-2,3,5,6-tetrafluorophenyl)porphyrine (2b)
A solution of 1b (1.97 g, 4.43 mmol) and pyrrole (0.29 g, 4.43 mmol) in dry dichloromethane (500 ml) was bubbled with argon for 30 min, BF3 x Et2O (0.17 ml, 1.33 mmol) was added dropwise, and reaction mixture was stirred at room temperature for 21 h. Then DDQ (1.11 g, 4.87 mmol) was added, and reaction mixture was refluxed for 2 h. After addition of Et3N (0.24 ml) the solvent was removed, residue was washed with cold methanol to uncolored solution. Product was purified in 2 steps: 1) flash-chromatography (silica gel, dichloromethane), 2) column chromatography (silica gel, hexane-chloroform = 1:1) to afford 0.23 g of violet powder (yield 21%). 1Н NMR (CDCl3, δ, ppm): -2.89 (s, 2H, NH); 5.07 (t, 8H, J = 12.87 Hz), 8.94 (s, 8H, Pyr-H). 19F NMR (CDCl3, δ, ppm): -78.51 (m, 8F, CAr-Forto); -59.44 (m, 8F, CAr-Fmeta); -48.45 (s, 8F, CF2), -46.20 (s, 8F, CF2); -42.93 (s, 8F, CF2), -3.04 (s, 12F, CF3). MS (APCI), m/z: 1895.4 [M+H]+.
meso-tetrakis(4-(pentafluorophenyloxy)-2,3,5,6-tetrafluorophenyl)porphyrine (2c)
A solution of 1c (0.72 g, 2 mmol) and pyrrole (0.18 g, 2,7 mmol) in dry dichloromethane (200 ml) was bubbled with argon for 30 min, BF3 x Et2O (0.1 ml, 0.8 mmol) was added dropwise, and reaction mixture was stirred at room temperature for 20 h. Then DDQ (0,61 g, 2,7 mmol) was added, and reaction mixture was refluxed for 2 h. After removing of the solvent product was purified by column chromatography (aluminium oxide, petroleum ether – chloroform = 6:1), washed with cold methanol to uncolored solution to afford 150 mg of violet powder (yield 18%). 1Н NMR (CDCl3, δ, ppm): -2.88 (s, 2H, NH); 8.96 (s, 8H, Pyr-H). 19F NMR (CDCl3, δ, ppm): -83.37 (t, 8F, J = 20.65 Hz, C6F5O (meta-F)); -80.95 (t, 4F, J 1= 22.95 Hz, J 2 = 20.65 Hz, C6F5O (para-F)); -78.66 (d, 8F, J = 18.36 Hz, C6F4-porph (meta-F)); -78.07 (d, 8F, J = 18.36 Hz, C6F5O (orto-F)); -58.80 (d, 8F, J = 13.77 Hz, C6F4-porph (orto-F)). Anal. calcd. for C68H10F36N4O4 (%): C, 50.08; H, 0.62; N, 3.44; F, 41.94. Found (%): C, 50.11; H, 0.49; N, 3.34; F, 41.65.
meso-tetrakis(2,4,6-tri(pentafluorophenyloxy)-3,5-difluorophenyl)porphyrine (2f)
A solution of 1f (0.68 g, 1 mmol) and pyrrole (0.06 g, 1 mmol) in dry chloroform (100 ml with addition of ethanol – 0.8 vol. %) was bubbled with argon for 30 min, BF3 x Et2O (0.05 ml, 0.4 mmol) was added dropwise, and reaction mixture was stirred at room temperature for 8 h. Then DDQ (0.23 g, 1 mmol) was added, and reaction mixture was stirred for 1.5 h. After addition of Et3N (0.05 ml) the solvent was removed, residue was washed with cold methanol to uncolored solution. Product was purified in 2 steps: 1) flash-chromatography (silica gel, chloroform), 2) column chromatography (silica gel, hexane-chloroform = 5:1) to afford 10 mg of violet powder (yield over 1%). 1Н NMR (CDCl3, δ, ppm): -3.44 (s, 2H, NH); 9.01 (s, 8H, Pyr-H). 19F NMR (CDCl3, δ, ppm): -85.65 (t, 16F, J1 = 18.31 Hz, J2 = 21.36 Hz, orto-OC6F5(meta-F)); -84.49 (t, 8F, J1 = 21.36 Hz, J2 = 18.31 Hz, para-OC6F5(meta-F)); -83.27 (t, 8F, J1 = 18.31 Hz, orto-OC6F5(para-F)); -81.14 (t, 4F, J = 21.36 Hz, para-OC6F5(para-F)); -79.80 (d, 16F, J = 18.31 Hz, orto-OC6F5(orto-F)), -78.55 (d, 8F, J = 21.36 Hz, para-OC6F5(orto-F)); -71.09 (s, 8F, C6F2-porph (meta-F)).
meso-tetrakis(4-(1,1-H,H-perfluoroheptyl-1-oxy)-2,3,5,6-tetrafluorophenyl)porphyrine (2g)
To a solution of meso-tetrakis(pentafluorophenyl)porphyrine (0.5 g, 0.51 mmol) and 1,1-H,H-perfluoroheptan-1-ol (2.16 g, 6.2 mmol) in THF (200 ml) KOH (2.86 g, 51 mmol) was added. Reaction mixture was refluxed under argon for 4 h. After removing of the solvent residue was poured into water and extracted by diethyl ether. Organic layer was washed with saturated aqueous solution of NaHCO3 (2 x 40 ml) and dried over CaCl2. After removing of the solvent product was purified by column chromatography (silica gel, chloroform) to afford 0.55 g of violet powder (yield 47%). 1Н NMR (CDCl3, δ, ppm): -2.86 (s, 2H, NH), 5.11 (t, 8H, J1 = 12.79 Hz, J2 = 12.56 Hz, OCH2), 8.97 (s, 8H, Pyr-H). 19F NMR (CDCl3, δ, ppm): -78.48 (m, 8F, CAr-Forto), -59.44 (dd, 8F, J1 = 13.79 Hz, J2 = 8.19 Hz, CAr-Fmeta), -48.38 (m, 8F, CF2), -45.14 (m, 8F, CF2), -44.33 (m, 8F, CF2), -42.73 (m, 8F, CF2), -2.98 (t, 12F, J = 9.05 Hz, CF3). Anal. calcd. for C72H18F68N4O4 (%): C, 35.00; H, 0.43; N, 2.00; F, 56.50. Found (%): C, 37.68; H, 0.79; N, 2.44; F, 56.30.
meso-tetrakis(4-(1,1-H,H-perfluorononyl-1-oxy)-2,3,5,6-tetrafluorophenyl)porphyrine (2h)
To a solution of meso-tetrakis(pentafluorophenyl)porphyrine (0.1 g, 0.1 mmol) and 1,1-H,H-perfluorononan-1-ol (1.2 g, 0.54 mmol) in THF (50 ml) K2CO3 (1.38 g, 10 mmol) was added. Reaction mixture was refluxed under argon for 12 h (reaction course was controlled by TLC). After removing of the solvent residue was poured into water and extracted by diethyl ether. Organic layer was washed with by saturated aqueous solution of NaHCO3 (2 x 40 ml) and dried over CaCl2. After removing of the solvent product was purified in 2 steps: 1) flash-chromatography (aluminium oxide, hexane – chloroform = 1:1), 2) column chromatography (aluminium oxide, gradient elution hexane - chloroform) to afford 95 mg of violet powder (yield 35%). 1H NMR (CDCl3, δ, ppm): -2.89 (s, 2H, NH), 5.06 (t, 8H, J = 12.72 Hz, OCH2), 8.94 (s, 8H, Pyr-H),. 19F NMR (CDCl3, δ, ppm): -78.47 (m, 8F, CAr-Forto), -59.43 (m, 8F, CAr-Fmeta), -48.34 (s, 8F, CF2), -45.17 (s, 8F, CF2), -44.92 (s, 8F, CF2), -44.07 (s, 24F, CF2CF2CF2), -42.65 (s, 8F, CF2), -3.00 (s, 12F, CF3). MS (APCI), m/z: 1834.6 [M+H-2C8F17CH2]+.
References
- DiMagno, S. G.; Dussault, P. H.; Schultz, J. A. J. Am. Chem. Soc. 1996, 118, 5312–5313.
- Pozzi, G.; Colombani, I.; Miglioli, M.; Montanari, F.; Quici, S. Tetrahedron 1997, 53, 6145–6162.
- Nakahara, H.; Liang, W.; Fukuda, K.; Wang, L.; Wada, T.; Sasabe, H. J. Colloid Interface Sci. 1998, 208, 14–22.
- Sun, H.; Smirnov, V. V; DiMagno, S. G. Inorg. Chem. 2003, 42, 6032–6040.
- Grancho, J. C. P.; Pereira, M. M.; Miguel, M. D. G.; Gonsalves, A. M. R.; Burrows, H. D. Photochem. Photobiol. 2002, 75, 249–256.
- Goll, J. G.; Moore, K. T.; Ghosh, A.; Therien, M. J. J. Am. Chem. Soc. 1996, 118, 8344–8354.
- Yi, W. Bin; Ma, J. J.; Jiang, L. Q.; Cai, C.; Zhang, W. J. Fluor. Chem. 2014, 157, 84–105.
- Gryko, D.; Wyrostek, D.; Nowak-Król, A.; Abramczyk, K.; Rogacki, M. Synthesis (Stuttg). 2008, 2008, 4028–4032.
- Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827–836.
- Lindsey, J.; Wagner, R. J. Org. Chem. 1989, 828–836.
- Preliminary results of this study have been published in the abstract book - X. German-Russian-Ukrainian Symposium on Fluorine Chemistry, Berlin, November 26-28, 2014, P.19 (Belyaeva, E.; Sigan, A.; Gervits, L.; Chkanikov N.). Subsequently, it turned that at the same time this approach have been investigated by Golf H. R. A. and co-workers and was published in A. European J. Org. Chem. 2015, 2015, 1548–1568. Reactions of TF20PPH2 with the number of fluorinated alkochols were considired there.
- Figueira, A. C. B.; de Oliveira, K. T.; Serra, O. A. Dye. Pigment. 2011, 91, 383–388.
- Hyun, M. Y.; Jo, Y. D.; Lee, J. H.; Lee, H. G.; Park, H. M.; Hwang, I. H.; Kim, K. B.; Lee, S. J.; Kim, C. Chem. - A Eur. J. 2013, 19, 1810.
Recommended for publication by Prof. S.R. Sterlin
Fluorine Notes, 2015, 102, 3-4
