Received: September, 2015
DOI 10.17677/fn20714807.2015.05.01
Fluorine Notes, 2015, 102, 1-2
Synthesis of New Floroaliphatic Functionalized Sulfonyl Bromides and Study of Their Chemical Properties
A.A. Tyutyunovab, L.F. Ibragimovaa, N.D. Kagramanova, N.I. Delyaginaa, V.F. Cherstkova, S.R. Sterlina, S.M. Igumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, V-334, GSP-1, 119991 Moscow, Russia
bNPO PiM-INVEST LLC, ul. Vavilova 28, 119991 Moscow, Russia
e-mail: tuytuynov@rambler.ru
Abstract: It is shown that fluoroaliphatic sulfonyl bromides containing terminal functional groups, such as alkoxycarbonyl, trifluorovinyl, or fluorosulfonyl, are convenient agents for radical fluoroalkylation of unsaturated hydrocarbons under photochemical initiation.
Keywords: tetrafluoroethane-β-sultone, ethyl bromodifluoroacetate, ethyl bromosulfonyl difluoroacetate, ethoxycarbonyldifluoromethylation, oxaperfluoroalkyl sulfonylbromides.
It was shown earlier that perfluoroalkyl sulfonylbromides RFSO2Br could be used for radical perfluoroalkylation of unsaturated and aromatic hydrocarbons [1-5]. Taking into consideration the literature data we assumed that sulfonylbromides derived from commercially available functionalized fluoroaliphatic bromides, e.g. bromosulfonyl difluoroacetates BrSO2CF2CO2R [R = Me (1a), Et (1b)], could be used as reagents for radical introduction of the corresponding fluoroaliphatic groups.
We failed to prepare bromosulfonyl difluoroacetylhalides 2 by the reaction of tetrafluoroethane-β-sultone (3) and anhydrous hydrogen bromide by analogy with synthesis of chlorosulfonyl difluoroacetylhalides [6]. The reaction between sultone 3 and anhydrous HBr in ether with the further treatment of the reaction mixture by dry methanol yielded the mixture of methyl bromodifluoroacetate (4) and disulfide 5 at the molar ratio of 2:1.
Scheme 1
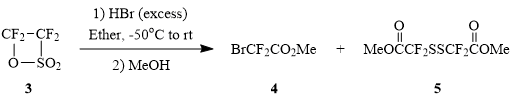
This reaction in CH2Cl2 results in formation of a complex mixture of products shown in Scheme 2:
Scheme 2
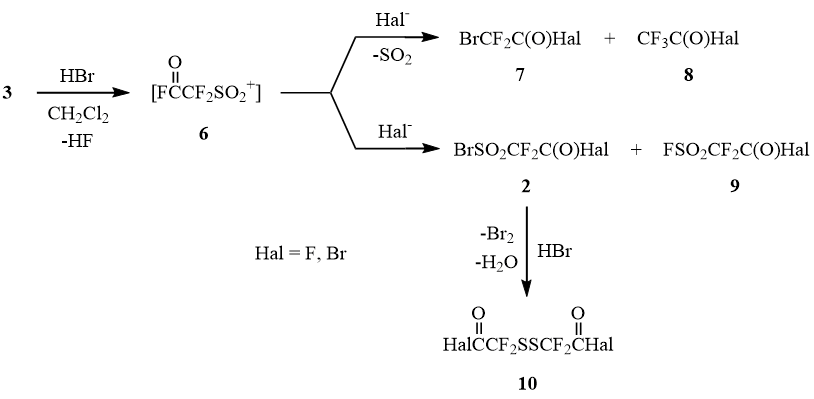
The difference in the composition of reaction products obviously reflects the effect of the solvent on the mechanism of the opening of sultone 3. In a nonpolar solvent, methylene chloride, the opening of sultone 3 occurs predominantly according to the electrophilic mechanism with formation of sulfonyl cation 6 that can both attach anions present in the reaction medium (F-, Br-) yielding products 2 and 9 and undergo desulfodioxidation with formation of acid halides of bromodifluoroacetic and trifluoroacetic acids 7 and 8. The assumed formation of disulfide 10 as a result of reduction of sulfonyl bromide 2 by hydrogen bromide was confirmed by high yield of 5 in the reaction between sultone 3 and AlBr3 in acetyl bromide medium.
Scheme 3

The suggested scheme is supported both by the considerable increase in the yield of sulfonyl bromides 2 (up to 40%) and by the absence of disulfide 10 among the products of the reaction between sultone 3 and HBr in the presence of bromine maintaining the oxidative medium, same as by the composition of the products of the reaction between 3 and AlBr3 in the medium of benzoyl bromide or dibromomethane, where acid halides 7 and 8 are predominant, but acid halides 2 and 10 are absent.
Formation of comparable amounts of compounds 4 and 5 in the course of the reaction between 3 and HBr in ether reflects in all probability the competition between the nucleophilic and electrophilic opening of the sultone: in the excess of ether HBr yields diethyloxonium bromide 11 that interacts with sultone 3 with intermediate formation of sulfonyl bromides 2 that are further quantitatively reduced to disulfides 10. At the same time, formation of acid halides of bromodifluoroacetic acid 7 is probably related to accumulation of water and hydrogen fluoride in the reaction mass, which results in generation of sulfonyl cation 6 that eliminates SO2 in the polar medium with the further formation of bromodifluoroacetyl halides 7.
Scheme 4

As follows from the above results, synthesis of derivatives of bromosulfonyl dufluoroacetic acid 2 on the basis of sultone 3 can hardly be considered as the preparative method of their synthesis. At the same time, it was shown earlier that perfluoroalkanesulfonyl halides are easily obtained by halogenation of the corresponding metal sulfinates formed in the reaction of sodium dithionite [1] or sulfurous anhydride in the presence of Zn, Al, Mn, or Cd with iodo- or bromoperfluoroalkanes [7].
Taking into account that methyl bromodifluoroacetate (4) and ethyl bromodifluoroacetate (12) are commercially available products, we attempted to synthesize ethyl bromosulfonyl difluoroacetate (1b) according to the method [7].
Indeed, it turned out that the reaction between ethyl bromodifluoroacetate 12 and Zn/SO2 in DMF with the further bromination of the reaction mixture resulted in formation of 1b with the yield of 45%.
Scheme 5
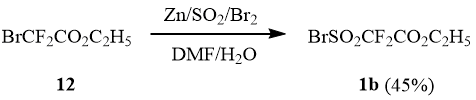
The suggested method of synthesis of sulfonyl bromide 1b, owing to availability of the initial bromodifluoroacetate 12, is preparatively convenient and can be easily carried out on a large scale, which to a certain extent compensates the low target product yield.
Sulfonyl bromide 1b is storage-stable in a fridge at 4-8°C; when heated (100°C) or under illumination by sunlight in a sealed ampoule, 1b undergoes desulfodioxidation by 4% in 4 h and by 5% in 10-12 h, accordingly.
The attempt to add sulfonyl bromide 1b to allyl trifluoroacetate (13) at the molar ratio of 1b:13 = 1:1 under the conditions of thermal initiation (100°C/4 h) resulted in predominant desulfodioxidation 1b with formation of a mixture of ester 12 and adduct 14a:
Scheme 6

The further studies showed that illumination of the mixture of 1b:13 at the molar ratio of 1:3 by sunlight in a sealed ampoule for 10-12 h results in formation of adduct 14a in the yield of 70%.
Scheme 7
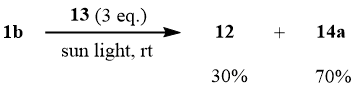
Adducts 14b-j of sulfonyl bromide 1b with other unsaturated hydrocarbon compounds were obtained in a similar way. The structure and yields of the obtained compounds are presented in Table 1.
Table 1. Reactions of ethyl bromosulfonyl difluoroacetate 1b with unsaturated compounds.
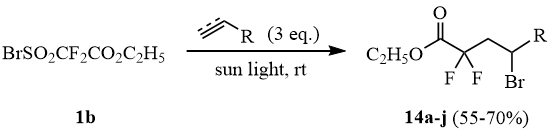
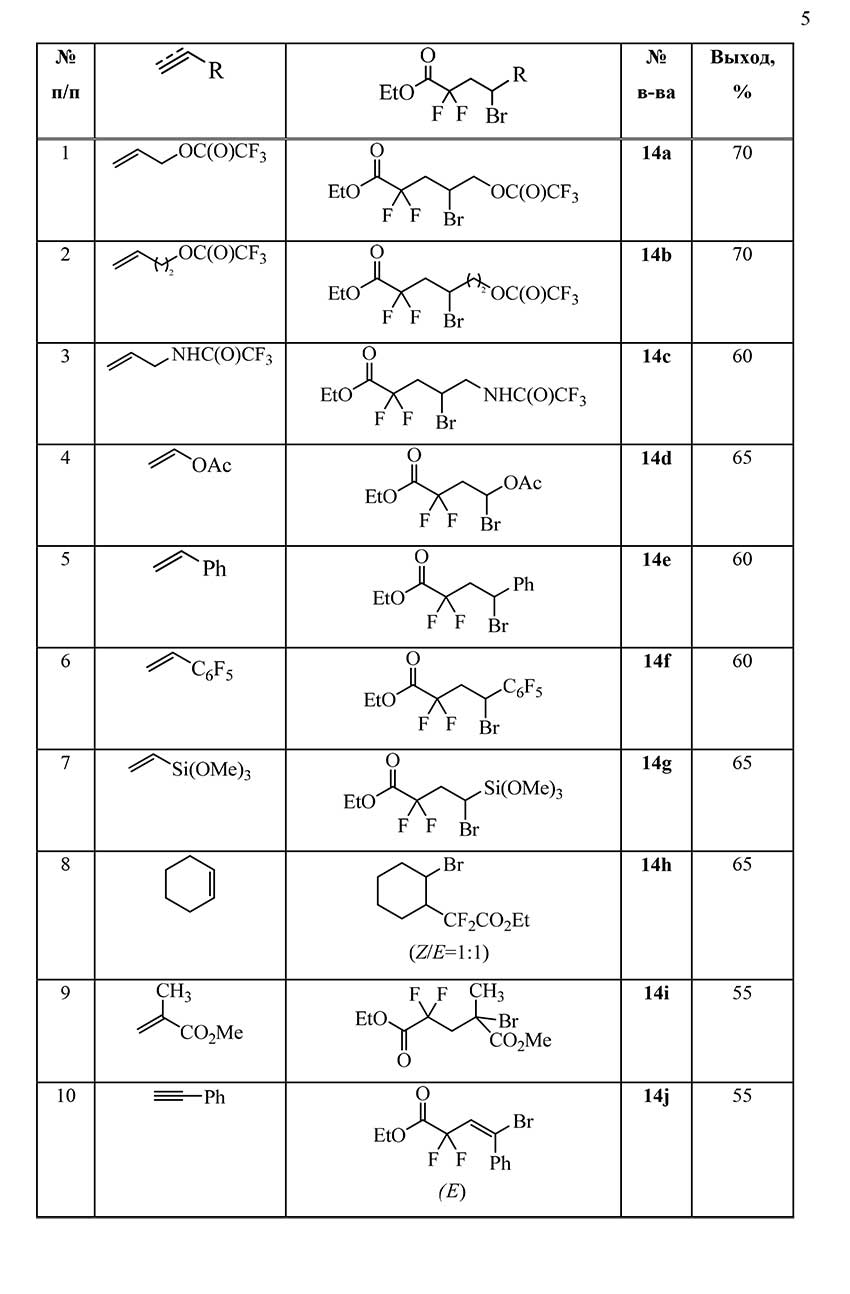
The performed experiments showed that ethyl bromosulfonyl difluoroacetate 1b adds to unsaturated compounds under mild conditions under exposure to sunlight and can be used for the embedding the ethoxycarbonyl difluoromethyl group into the hydrocarbon chain.
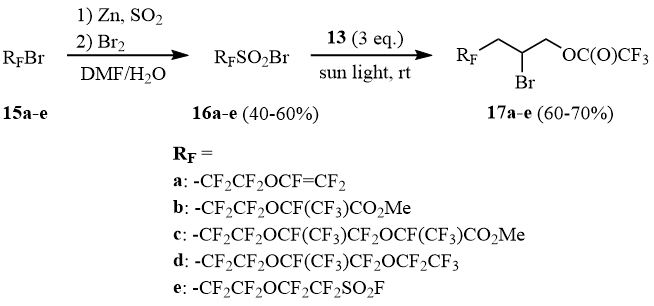
Reactivity close to that of sulfonyl bromide 1b is also specific for sulfonyl bromides 16a-e obtained by the reaction of primary fluoroaliphatic bromides 15a-e with Zn/SO2 with the further bromination of the forming zinc sulfinates (Scheme 8). Sulfonyl bromides 16a-e adds to allyl trifluoroacetate (13) under exposure to sunlight and form adducts 17a-e in 60-70% yield:
Scheme 8
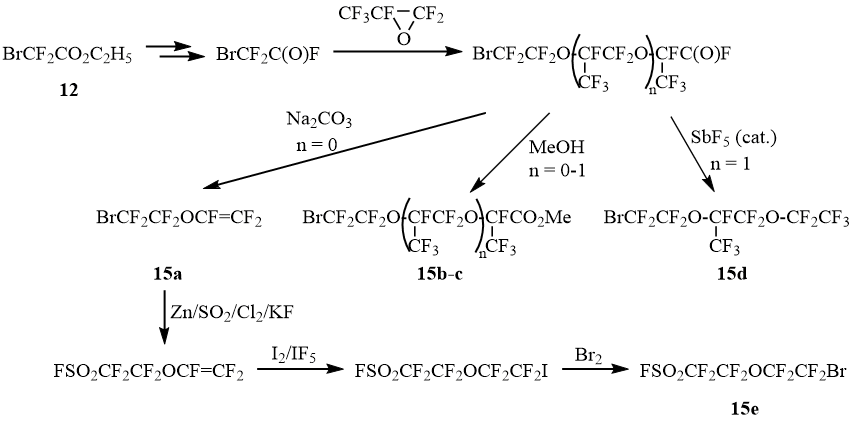
The initial bromides 15a-e are obtained on the basis of bromodifluoroacetate 12 through a series of transformations shown in Scheme 9 [8-11]:
Scheme 9
As opposed to sulfonyl bromides 1a-b and 16b-e that undergo desulfodioxidation under exposure to sunlight with formation of bromides 12 and 15b-e, sulfonyl bromide 16a yields in sunlight a mixture of bromide 15a and cyclic bromide 18:
Scheme 10
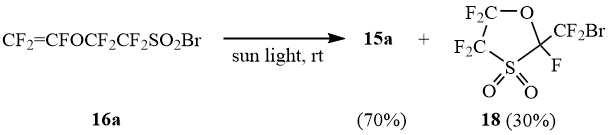
Thus the embedding of SO2 across C-Br bond in fluoroaliphatic bromides can be considered as a latent form of its activation, which allows to compensate the relitavely low reactivity of fluoroaliphatic bromides in the reactions of radical addition.
Experimental
1H, 19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300 and 282 MHz, accordingly; the external standard was CDCl3. Chemical shifts for 1H spectra are presented vs. the residual signal of the solvent (δ 7.26) and are given in ppm vs. tetramethylsilane. Chemical shifts in 19F spectra are given in ppm vs. CFCl3. Downfield shifts are positive. Raman spectra are recorded on a Jobin Yvon LabRam spectrometer. Mass spectra are recorded using a Finnigan Polaris Q mass spectrometer (Trace GC ultra).
The method of synthesis of sultone 3 has been described earlier in [12-13].
Reaction of Tetrafluoroethane-β-Sultone (3) with HBr in Ether.
Tetrafluoroethane- β-sultone (3) (14.55 g, 0.081 mol) is added dropwise to the ether solution of dry HBr (18.36 g, 0.227 mol, in 100 g of dry ether) under stirring at the temperature of -50÷-45°C. Then, the temperature of the reaction mixture is increased to 20÷25°C and the mixture is stirred at this temperature for 30 min. Further, the mixture is cooled to -20÷-15°C and dry methanol (15 ml) is added under stirring; the mixture temperature is increased to 20÷25°C and the mixture is stirred at this temperature for 30 min. Then the volatile components are distilled in vacuum (10-15 torr) into a trap (-78°C). The obtained liquid residue (10.6 g) is analyzed using 1H, 19F NMR.
19F NMR δ: -84, s, (5) (cf. [14]), -62, s, (4), the molar ratio of 5:4=1:2.
The obtained mixture is twice washed with cold water; the lower layer contains, according to 1H and 19F NMR data, the esters 5 and 4. The mass spectrum of bis(methoxycarbonyl difluoromethyl)disulfide (5) (M/Z, reference): 297[M+CH3]+, 282[M]+, 263[M-F]+, 235[C5H3F4O2S2]+, 218[M-2S]+(100%), 195[C5H6FO2S2]+, 181[C5H6FO4S]+, 154[C3H3FO2S2]+, 141[C3HF2OS]+, 124[C2HFOS2]+, 121[C3H2FO2S]+, 114[CF2S2]+, 109[C3F3O]+, 93[C3F3]+, 82[CF2S]+, 81[C2F3]+, 64[S2]+, 63[CFS]+, 59[C2H3O2]+, 45[CHS]+.
Reaction of Tetrafluoroethane-β-Sultone (3) with HBr in CH2Cl2.
Tetrafluoroethane- β-sultone (3) (18 g, 0.1 mol) is added dropwise to the solution of anhydrous HBr (24 g, 0.3 mol) in CH2Cl2 (100 ml) at -50oC; at the end of addition, the reaction mixture is stirred for 30 min, then dry methanol is added dropwise; the whole is heated to the room temperature, the reaction mass is poured onto crashed ice, the organic layer is separated, washed with dilute aqueous NaHCO3 solution to give 12.4 g of a mixture of esters 1a, 4, 5, FSO2CF2CO2Me, and CF3CO2Me identified by GLC, 19F NMR and chromato-mass spectrometry methods.
Reaction of Tetrafluoroethane-β-Sultone (3) with AlBr3 in CH3C(O)Br.
Tetrafluoroethane- β-sultone (3) (18 g, 0.1 mol) is added dropwise to the solution of aluminum bromide (5.4 g, 0.02 mol) in acetyl bromide (75 g, 0.61 mol) at -50oC, stirred for 30 min, then dry methanol is added dropwise; the whole is heated to 20÷25oC, the reaction mass is poured onto ice, the organic layer is separated, washed with dilute aqueous NaHCO3 solution to dive 12.7 g of ester 5 (90%).
Sulfodioxidation of Ethyl Bromodifluoroacetate (12).
Ethyl Bromosulfonyldifluoroacetate (1b).
Zn powder (14.65 g, 0.224 mol) and water (4 ml) are added under stirring to the solution of ethyl bromodifluoroacetate (12) (100 g, 0.49 mol) and SO2 (57.4 g, 0.896 mol) in DMF (350 ml) cooled to 5°C; upon that, the mixture temperature increases to 35÷45°C. The reaction mixture is cooled to 25÷30°C, stirred at this temperature for 1 h, cooled to 10÷15°C, Zn powder (14.65 g, 0.224 mol) and water (4 ml) are added; meanwhile, the mixture temperature rises to 30÷35°C. Then the reaction mixture is cooled to 25÷30°C, stirred at this temperature for 3 h, cooled to -15÷-10°C and bromine (115 g, 0.72 mol) is added dropwise under stirring. The temperature mixture is increased to 5÷10°C; the reaction mixture is poured into ice water (500 ml), hydrochloric acid (100 ml) is added under stirring, the lower layer is separated, washed with dilute hydrochloric acid and distilled over P2O5 in vacuum; the fraction at 55-115oC/10 torr is collected.
Further rectification yields 47 g (45% with account for conversion of 12), b.p. 65-66°C/1.5 torr. Found (%): C, 17.93; H, 1.90; B, 30.18; F, 14.20; S, 11.78. C4H5BrF2O4S. Calculated (%): C, 17.99; H, 1.89; B, 29.92; F, 14.23; S, 12.01. 1H NMR δ: 1.6 (t, 3H, CH3), 4.7 (q, 2H, CH2); 19F NMR δ: -101 (s, CF2). The mass spectrum (M/Z, reference): 267[M+H]+, 239[C2H2BrF2O4S]+, 203[C4H6BrF2O2]+, 175[C3H6BrF2O]+, 59[C2H3O2]+(100%), 51[CF2H]+, 29[C2H5]+.
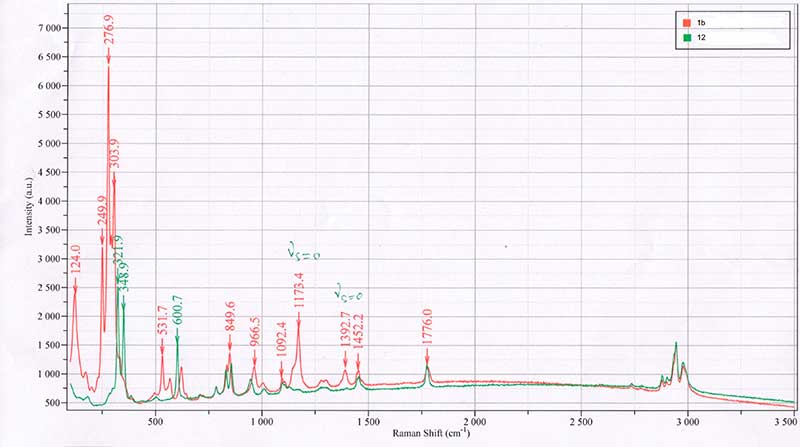
Fig. 1. Raman spectra of compounds 1b and 12.
Synthesis of Compounds 14a-j (Typical Experiment).
A mixture of ethyl bromosulfonyldifluoroacetate (1b) (2 g, 7.5 mmol) and olefin or phenyl acetylene (22.5 mmol) (Table 1) is placed into a tube of molybdenum glass (the loading factor is 10%) sealed by a stopper and conditioned for 24 h under sunlight irradiation (the temperature is 25-30°C, cloudy weather produces no significant effect on the reaction).
Volatile mixture components are evacuated at 25÷50°C/15-0.1 torr into a trap (-78°C) to give a residue containing adducts 14a-j. The yields are presented in Table 1.
Ethyl 4-bromo-2,2-difluoro-5-(2,2,2-trifluoroacetoxy)valerate (14a).
B.p. 124-125оС/10 torr. Found (%): C, 30.24; H, 2.94; F, 26.49. C9H10BrF5O4. Calculated (%): C, 30.27; H, 2.82; F, 26.60. 1H NMR δ: 1.3 (t, 3H, CH3), 2.8 (dt, 2H, CH2CF2), 4.3 (q, 2H, CH2CH3), 4.4 (m, 1H, CHBr), 4.6 (d, 2H, CH2O); 19F NMR δ: -108, -104 (ABq, 2F, 2JFF = 282 Hz, CF2CO2Et), -77 (s, 3F, CF3). The mass spectrum (M/Z, reference): 357[M+H]+, 277[M-Br]+, 249[C7H6F5O4]+, 243[C7H10BrF2O2]+, 215[C6H10BrF2O]+, 195[C5H2BrF2O]+, 169[C4H4BrF2]+, 163[C7H9F2O2]+, 135[C5H5F2O2]+(100%), 115[C5H4FO2]+, 107[C4H5F2O]+, 91[C4H5F2]+, 90[C4H4F2]+, 71[C4H4F]+, 69[CF3]+, 51[CF2H]+.
Ethyl 4-bromo-2,2-difluoro-5-(2,2,2-trifluoroacetoxy)caproate (14b).
B.p. 145-147оС/10 torr. 1H NMR δ: 1.3 (t, 3H, CH3), 2.1-2.5 (m, 2H, CH2CH2O), 2.6-3 (m, 2H, CH2CF2), 4.3 (m, 3H, CHBr, CH2CH3), 4.6 (m, 2H, CH2O); 19F NMR δ: -107.7, -103.3 (ABq, 2F, 2JFF = 282 Hz, CF2CO2Et), -77 (s, 3F, CF3). The mass spectrum (M/Z, reference): 371[M+H]+, 370[M]+, 291[M-Br]+, 263[C9H12F5O3]+, 177[C8H11F2O2]+, 149[C6H7F2O2]+(100%), 129[C6H3F2O]+, 103[C5H7F2]+, 69[CF3]+.
Ethyl 4-bromo-2,2-difluoro-5-(2,2,2-trifluoroacetamido)valerate (14c).
1H NMR δ: 1.4 (t, 3H, CH3CH2), 2.85 (dt, 2H, CH2CF2), 3.85 (m, 1H, CHBr), 4 (m, 2H, CH2NH) 4.4 (q, 2H, CH3CH2), 8.5 (m, 1H, NH); 19F NMR δ: -107.95, -103.05 (АВq, 2F, 2JFF = 282 Hz, CF2CO2Et), -77.5 (s, 3F, CF3). The mass spectrum (M/Z, reference): 356[M+H]+, 276[C9H11F5O3N]+, 248[C8H11F5O2N]+, 230[C7H5F5O3N]+, 214[C7H5BrO2N]+, 202[C6H3F5ON]+(100%), 182 [C6H2F4ON]+, 163[C6H2F3ON]+, 135[C4H4F3]+, 126[C3H3F3ON]+, 112[C2HF3ON]+, 69[CF3]+, 51[CHF2]+.
Ethyl 4-acetoxy-4-bromo-2,2-difluorobutyrate (14d).
B.p. 125-127оС/10 torr. 1H NMR δ: 1.5 (t, 3H, CH3CH2), 2.25 (s, 3H, CH3), 3.1-3.6 (m, 2H, CH2CF2), 4.6 (q, 2H, CH3CH2), 7 (m, 1H, CHBr); 19F NMR δ: -107.4, -105.6 (АВq, 2F, 2JFF = 282 Hz, CF2CO2Et). The mass spectrum (M/Z, reference): 289[M+H]+, 209[C8H10F2O4]+, 167[C6H8F2O3]+(100%), 147[C6H7FO3]+, 51[CHF2]+, 43[C2H3O]+.
Ethyl 4-bromo-2,2-difluoro-4-phenylbutyrate (14e).
1H NMR δ: 0.9 (t, 3H, CH3CH2), 2.8-3.1 (m, 2H, CH2CF2), 3.75 (q, 2H, CH3CH2), 5 (m, 1H, CHBr), 6.9-7.2 (m, 5H, C6H5); 19F NMR δ: -105.23, -104.57 (АВq, 2F, 2JFF = 282 Hz, CF2CO2Et). The mass spectrum (M/Z, reference): 227[M-Br]+, 207[C12H12FO2]+, 187[C12H11O2]+, 169[C7H6Br]+, 159[C10H4FO]+, 153[C9H7F2]+, 133[C9H6F]+, 131[C6H8FO2]+(100%), 115[C9H7]+, 104[C8H8]+, 103[C8H7]+, 51[CHF2]+.
Ethyl 4-bromo-2,2-difluoro-4-(pentafluorophenyl)butyrate (14f).
B.p. 95оС/1.5 torr. 1H NMR δ: 1.1 (t, 3H, CH3), 2.8-3.1 (m, 2H, CH2CF2), 4 (q, 2H, CH3CH2), 5.3 (m, 1H, CHBr); 19F NMR δ: -164 (m, 2F, m-FC6F5), -156 (m, 1F, p-FC6F5), -143 (m, 2F, o-FC6F5), -109.4, -106.6 (АВq, 2F, 2JFF = 282 Hz, CF2CO2Et). The mass spectrum (M/Z, reference): 397[M+H]+, 317[M-Br]+(100%), 297[C12H7F6O2]+, 271[C10H2F7O]+, 269[C10F7O]+, 259[C7HBrF5]+, 249[C11H6F5O]+, 241[C7H2BrF4]+, 221[C9F6]+, 194[C8H3F5]+, 174[C8H2F4]+, 161[C7F4]+, 143[C7HF3]+, 105[C4H3F2O]+, 51[CHF2]+.
Ethyl 4-bromo-2,2-difluoro-4-(trimethoxysilyl)butyrate (14g).
1H NMR δ: 1.4 (t, 3H, CH3СН2), 2.6-2.9 (m, 2H, CH2CF2), 3.4 (m, 1H, CHBr), 3.75 (s, 9Н, Si(OCH3)3), 4.4 (q, 2H, CH3CH2); 19F NMR δ: -108.95, -104.05 (АВq, 2F, 2JFF = 282 Hz, CF2CO2Et). The mass spectrum (M/Z, reference): 319[C8H14BrF2O4Si]+(100%), 299[C8H13BrFO4Si]+, 271[C9H17F2O5Si]+, 251[C9H16BrFO5Si]+, 122[C5H2O2Si]+, 103[C4HF2O]+, 91[C3HF2O]+.
(Z, E)-Ethyl (2-bromocyclohexyl)-2,2-difluoroacetate (14h).
1H NMR δ: 1.25 (m, 3H, CH3СН2), 1.6-2.6 (m, 8H, Су), 4 (dt, 1H, CHCF2), 4.2-4.3 (m, 2H, CH3CH2), 4.5 (s, 1H, CHBr); 19F NMR δ: -116.5, -107.5 (АВq, 2F, 2JFF = 282 Hz, Z-isomer), -111.31, -110.19 (АВq, 2F, 2JFF = 282 Hz, CF2CO2Et, E-isomer). The mass spectrum (M/Z, reference): 285[M+H]+, 233[C8H7BrFO2]+, 205[M-Br]+(100%), 185[C10H14FO2]+, 177[C8H11F2O2]+, 157[C8H10FO2]+, 131[C7H9F2]+, 109[C6H2FO]+, 91[C6H3O]+, 81[C6H9]+, 77[C5HO]+, 51[CHF2]+.
(E)-Ethyl 4-bromo-2,2-difluoro-4-phenylbut-3-enoate (14j).
B.p. 110/1 torr. 1H NMR δ: 0.9 (t, 3H, CH3CH2), 3.8 (q, 2H, CH3CH2), 6.5 (t, 1H, CHCF2), 7.15 (m, 3H, m,p-HC6H5), 7.3 (m, 2H o-HC6H5); 19F NMR δ: -94.5 (d, 2F, 3JFH = 11 Hz, CF2CO2Et). The mass spectrum (M/Z, reference): 305[M+H]+, 285[M-F]+, 265[C12H10BrO2]+, 231[C9H6BrF2]+, 225[C12H11F2O2]+, 213[C9H7BrF]+(100%), 197[C11H11F2O]+, 169[C9H7F2O]+, 151[C9H5F2]+, 133[C9H6F]+, 102[C8H6]+, 91[C3HF2O]+, 76[C3H2F2]+, 75[C3HF2]+, 63[C2HF2]+, 50[CF2]+.
General Method of Synthesis of Sulfonyl bromides 16a-c and 16e.
Zn powder (9.8 g, 0.15 mol) and water (2.7 ml) are added under stirring to the solution of bromide 15a-c or 15e (0.33 mol) and SO2 (38.4 g, 0.6 mol) in DMF (300 ml) cooled to 5°C; upon that, the mixture temperature increases to 35÷45°C. The reaction mixture is cooled to 25÷30°C; stirred at this temperature for 1 h, cooled to 10÷15°C, Zn powder (9.8 g, 0.15 mol) and water (4 ml) are added; meanwhile, the mixture temperature rises to 30÷35°C. Then the reaction mixture is cooled to 25÷30°C, stirred for 3 h, cooled to -15÷-10°C and bromine (76.8 g, 0.48 mol) is added dropwise under stirring, the temperature is increased to 5÷10°C. The reaction mixture is poured into ice water (500 ml), hydrochloric acid (100 ml) is added under stirring, the lower layer is separated, washed with dilute hydrochloric acid and distilled over P2O5 in vacuum; further rectification is used to separate sulfonyl bromides 16a-c and 16e.
2-(Trifluorovinyloxy)tetrafluoroethane sulfonyl bromide (16a).
Yield 60%, b.p. 33-34оС/10 torr. Found (%): C, 14.10; F, 39.04; S, 9.05. C4BrF7O3S. Calculated (%): C, 14.09; F, 39.00; S, 9.40. 19F NMR δ: -137.1 (dd, 1F, 3JFF-trans = 112 Hz, 3JFF-cis = 68 Hz, OCF), -123.3 (dd, 1F, 2JFF = 90 Hz, =CF-trans), -115.8 (dd, 1F, 2JFF = 90 Hz, =CF-cis), -109.9 (s, 2F, CF2SO2Br), -82.9 (s, 2F, OCF2).
Methyl 3-oxa-5-bromosulfonylperfluoro-2-methylvalerate (16b).
Yield 60%, b.p. 52оС/1 torr. 1H NMR δ: 4.1 (s, 3H, CH3); 19F NMR δ: -133.1 (m, 1F, CF), -110.2 (s, 2F, CF2SO2Br), -84.2 (s, 3F, CF3), -83.4, -75.2 (АВq, 2F, 2JFF = 143 Hz, CF2O).
Methyl 3,6-dioxa-8-bromosulfonylperfluoro-2,5-dimethylcaprylate (16c).
Sulfonylbromide was synthesized according to the general procedure in 450 ml of DMF per 0.33 mol of ester 15c. The rectification of organic layer resulted in separation of a fraction (70-95oC/0.5 torr) that contained 70% 16c and 30% 15c, yield 50% (based on 15c entered the reaction). 1H NMR spectrum of 16c δ: 4.0 (s, 3H, CH3); 19F NMR δ: -146.5 (m, 1F, CFCF2), -133.6 (m, 1F, CFCO2Me), -110.3 (s, 2F, CF2SO2Br), -84.7 (m, 6F, CF3), -88÷-80 (m, 2F, CFCF2O), -78 (m, 2F, CF2CF2O).
3,6-Dioxaperfluoro-4-methyloctane Sulfonyl Bromide (16d).
Zn powder (9.8 g, 0.15 mol) and water (2.7 ml) are added under stirring to the solution of SO2 (38.4 g, 0.6 mol) in DMF (450 ml) cooled to 5°C; upon that, the mixture temperature increases to 35÷45°C. The reaction mixture is cooled to 10÷15°C; Zn powder (9.8 g, 0.15 mol) and water (2.7 ml) are added; meanwhile, the mixture temperature rises to 30÷35°C. Then bromide 15d (96.2 g, 0.2 mol) is added into the reaction mixture, the whole is stirred vigorously at 55÷60°C for 3-4 h, cooled to -15÷-10°C and bromine (76.8 g, 0.48 mol) is added dropwise under stirring. The temperature mixture is increased to 5÷10°C; the reaction mixture is poured into ice water (500 ml), hydrochloric acid (100 ml) is added under stirring, the lower layer is separated, washed with dilute hydrochloric acid and distilled over P2O5 in vacuum; further rectification is used to separate sulfonyl bromide 16d.
Yield 60%, b.p. 57-58оС/15 torr. 19F NMR δ: -147.1 (m, 1F, CF), -110.7 (s, 2F, CF2SO2Br), -90.9 (m, 2F, CF2O), -89.7 (m, 3F, CF3), -85.2 (m, 2F, CF2O), -82.4 (m, 3F, CF3), -78.4 (m, 2F, CF2O).
3-Oxa-5-bromosulfonylperfluoropentane sulfonyl fluoride (16e).
Yield 40%. b.p. 72-73оС/15 torr. ЯМР 19F δ: -114.1 (m, 2F, CF2SO2F), -110.4 (s, 2F, CF2SO2Br), -83.5 (m, 2F, CF2O), -80.7 (m, 2F, CF2O), 43.9 (m, 1F, SO2F).
The synthesis of compounds 17a-e.
The adducts 17a-e are prepared from sulfonylbromides 16a-e and allyl trifluoroacetate (13) under the conditions of preparing compounds 14a-j.
2-Bromo-4,4,5,5-tetrafluoro-5-trifluorovinyloxypentyl trifluoroacetate (17a).
Yield 70%. 1H NMR δ: 2.75 (m, 2H, CH2CF2), 4.4 (m, 1H, CHBr), 4.6 (d, 2H, CH2O); 19F NMR δ: -137.6 (dd, 1F, 3JFF-trans = 112 Hz, 3JFF-cis = 68 Hz, OCF), -125.2 (dd, 1F, 2JFF = 90 Hz, =CF-trans), -118.9 (m, 2F, CF2), -117.3 (dd, 1F, 2JFF = 90 Hz, =CF-cis), -92.5 (s, 2F, CF2O), -77.5 (s, 3F, CF3). The mass spectrum (M/Z, reference): 351[M-Br]+, 333[C7H5BrF7O2]+, 313[C7H4BrF6O2]+, 297[C7H4BrF6O]+, 279[C7H3BrF5O]+, 269[C7HBrF3O3]+, 249[C7BrF2O3]+, 219[C5H4BrF4]+, 199[C5H3BrF3]+, 189[C5H5F4O3]+, 169[C4H4BrF2]+, 157[C5H5F4O]+, 155[C3H2BrF2]+, 145[C4H5F4O]+, 139[C5H3F4]+, 109[C4H4F3]+, 95[C3H5F2O]+, 77[C3H3F2]+, 69[CF3]+(100%), 51[CHF2]+.
Methyl 3-oxa-7-bromo-8-trifluoroacetoxy-2-trifluoromethyl-2,4,4,5,5-pentafluorocaprilate (17b).
Yield 70%. 1H NMR δ: 2.8 (m, 2H, CH2CF2), 3.95 (s, 3H, CH3), 4.5 (m, 1H, CHBr), 4.65 (m, 2H, CH2O); 19F NMR δ: -134.1 (m, 1F, CF), -118.8 (m, 2F, CF2CH2), -93.4, -86.0 (АВq, 2F, 2JFF = 143 Hz, CF2O), -84.7 (s, 3F, CFCF3), -77.5 (s, 3F, CF3CO2). The mass spectrum (M/Z, reference): 523[M+СН3]+, 449[C9H5BrF11O3]+, 429[M-Br]+, 409[C11H7F10O5]+, 375[C10H4BrF11O4]+, 355[C10H3BrF10O4]+, 335[C10H2BrF9O4]+, 315[C9H7F8O3]+, 295[C9H6F7O3]+(100%), 275[C9H5F6O3]+, 233[C7H4F6O2]+, 199[C5H3BrF3]+, 169[C4H4BrF2]+, 151[C4H5BrF]+, 131[C4H4Br]+, 89[C4H3F2]+, 69[CF3]+, 59[C2H3O2]+.
Methyl 3,6-dioxa-10-bromo-11-trifluoroacetoxy-2,5-di(trifluoromethyl)-2,4,4,5,7,7,8,8-octafluoroundecanoate (17c).
Mixture of stereoisomers, yield 60%. 1H NMR δ: 3.1 (m, 2H, CH2CF2), 4.2 (m, 3H, CH3), 4.7 (m, 1H, CHBr), 4.9 (m, 2H, CH2O); 19F NMR δ: -147.1 (m, 1F, CFCF2), -133.5 (m, 1F, CFCO2Me), -118.8 (m, 2F, CF2CH2), -88÷-80 (m, 4F, CF2OCFCF2O), -84.5 (s, 3F, CF3), -81.8 (s, 3F, CF3), -77.4 (m, 3F, CF3CO2). The mass spectrum (M/Z, reference): 595[M-Br]+, 540[C12H6BrF13O4]+, 521[C12H6BrF12O4]+, 461[C12H6F13O4]+, 441[C12H5F12O4]+, 325[C7H3F10O3]+, 297[C9H5F8O2]+, 199[C5H3BrF3]+, 150[C3F6]+, 131[C3F5]+(100%), 119[C5H2F3]+, 81[C2F3]+, 69[CF3]+, 59[C2H3O2]+, 51[CHF2]+, 39[C3H3]+.
6,9-Dioxa-2-Bromo-7-trifluoromethyl-4,4,5,5,7,8,8,10,10,11,11,11-dodecafluoroundecyl trifluoroacetate (17d).
Yield 65%. 1H NMR δ: 3.1 (m, 2H, CH2CF2), 4.9 (m, 1H, CHBr), 5.1 (m, 2H, CH2O); 19F NMR δ: -147.1 (m, 1F, CF), -118.9 (m, 2F, CF2CH2), -90.5 (m, 2F, CF2O), -89.1 (s, 3F, CF3), -87.6 (m, 2F, CF2O), -84.9 (s, 2F, CF2O), -82.0 (m, 3F, CFCF3), -77.4 (s, 3F, CF3CO2). The mass spectrum (M/Z, reference): 615[M-F]+, 555[M-Br]+, 521[C10H5BrF15O2]+, 501[C10H5BrF14O2]+, 419[C10H2F14O2]+, 255[C7H3F8O]+, 219[C5H4BrF4]+, 199[C5H3BrF3]+, 169[C4H4BrF2]+, 155[C3H2BrF2]+, 139[C5H3F4]+, 119[C5H2F3(C2F5)]+, 95[C3H2F3]+, 89[C4H3F2]+, 77[C3H3F2]+, 69[CF3]+(100%), 51[CHF2]+.
3-Oxa-7-bromo-8-trifluoroacetoxy-1,1,2,2,4,4,5,5-octafluorooctane sulfonyl fluoride (17e).
Yield 60%. 1H NMR δ: 3.1 (m, 2H, CH2CF2), 4.9 (m, 1H, CHBr), 5.1 (m, 2H, CH2O); 19F NMR δ: -118.9 (m, 2F, CF2CH2), -114.1 (s, 2F, CF2SO2F), -89.7 (m, 2F, CF2O), -83.9 (m, 2F, CF2O), -77.2 (s, 3F, CF3CO2), 43.2 (m, 1F, SO2F). The mass spectrum (M/Z, reference): 453[M-Br]+, 418[C7H4BrF9O3S]+, 398[C7H3BrF8O3S]+, 379[C7H3BrF7O3S]+, 335[C7H4BrF8O]+, 317[C7H4BrF7O]+, 273[C7H5F8O2]+, 253[C7H4F7O2]+, 233[C7H3F6O2]+, 213[C7H2F5O2]+, 199[C5H3BrF3]+, 183[C2F5O2S]+, 169[C4H4BrF2]+, 155[C3H2BrF2]+, 139[C5H3F4]+(100%), 119[C5H2F3]+, 117[C3H2Br]+, 109[C4H4F3]+, 100[C2F4]+, 95[C3H2F3]+, 89[C4H3F2]+, 69[CF3]+, 67[C4F]+, 51[CHF2]+, 39[C3H3]+.
2,4,4,5,5-Pentafluoro-2-(bromodifluoromethyl)-3-oxathiolane-1,1-dioxide (18).
19F NMR δ: -128.1, -125.2 (ABq, 2F, 2JFF = 214 Hz, CF2SO2), -126.8 (s, 1F, CFO), -87.4, -83.3 (ABq, 2F, 2JFF = 90 Hz, CF2O), -68.2, -67.3 (ABq, 2F, 2JFF = 14 Hz, CF2Br).
References
- W.-Y. Huang J.Fluor.Chem., 1992, 58, 1-8.
- W.-Y. Huang, J.-L. Chen ActaChim.Sinica, Engl.Ed., 1986, 4, 381-386.
- W.-Y. Huang, J.-L. Chen ActaChim.Sinica, Engl.Ed., 1988, 6, 150-154.
- Y.-F. Zhang, L. Lu, W.-Y. Huang ActaChim.Sinica, Engl.Ed., 1989, 7, 376-384.
- W.-Y. Huang, H.-Z. Zhang Chin.J.Chem., 1991, 9, 76-83.
- G.A. Sokol’skii, M.A. Belaventsev, I.L. Knunyants Bull.Acad.Sci.USSR, Div.chem.sci., 1967, 16, 1471-1474.
- C. Wakselman J.Fluor.Chem., 1992, 59, 367-378.
- S.M. Igumnov, G.I. Lekontseva, A.A. Shipigusev, V.F. Mukhametshin Russ.J.Appl.Chem., 2005, 78, 435-437.
- S.M. Igumnov, S.R. Sterlin, A.A. Tjutjunov, Z.A. Mikhajlova Patent RU N 2497801 (2013).
- S.M. Igumnov, S.R. Sterlin, A.A. Tjutjunov Patent RU N 2503659 (2014).
- S.M. Igumnov, A.A. Tjutjunov Patent RU N 2475477 (2013).
- D.C. England, H. Oak U.S. Patent №2,852,554 (1958).
- I.L. Knunyants, M.A. Dmitriev, G.A. Sokol’skii USSR Certificate of Authorship N 116578 (1958).
- W.R. Brasen, H.N. Cripps, C.G. Bottomley, M.W. Farlow, C.G. Krespan J.Org.Chem., 1965, 30, 4188-4193.
Recommended for publication by Prof. S.R. Sterlin
Fluorine Notes, 2015, 102, 1-2
