Received: March, 2015
DOI 10.17677/fn20714807.2015.04.02
Fluorine Notes, 2015, 101, 3-4
Structural Properties,Theory Functional Calculations (DFT), Natural Bond Orbital and Energies for the fluorocarbon compounds
Shahriar Ghammamy, Farzane Yousefi
Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin,
Iran
e-mail: shghamamiii@yahoo.com or
farzaneyousefi@yahoo.com
Abstract: The structural and electronic properties of wurtzite C20H11F13ClN3O3S structure have been theoretically investigated within the framework of the density functional theory method(1). B3LYP/3-21G calculation results indicated that some selected bond length and bond angles values for the C20H11F13ClN3O3S In this paper, the optimized geometries and frequencies of the stationary point and the minimum-energy paths of two new compounds with C20H11F13ClN3O3S chemical formula are calculated by using the DFT methods with 3-21G basis set. The detail group points of compound is C1.
Keywords: Electronic structure, Fluorocarbon compounds, DFT Calculations, Vibrational analysis, B3LYP level.
1. Introduction
Sulfa drugs are still today among the drugs first used (together with ampicillin and gentamycin)as chemotherapeutic agents in bacterial infections by Escherichia coli in humans (2).Sulfonamides are antimicrobial agents widely employed in animal production and their residues in food could be an important risk to human health. In the dairy industry, large quantities of milk are monitored daily for the presence of sulfonamides. Fluoroalkanes can serve as oil-repellent/water-repellent fluoropolymers, solvents, liquid breathing research agents, and powerful greenhouse gases. Fluorocarbon liquids are colorless. They have high density, up to over twice that of water, due to their high molecular weight. Recently, perfluorochemicals (PFCs) are of specialpublic concern as environmental contaminantsbecause they are globally distributed, persistent, andbioaccumulative for higher chain homologues. During this study we report the optimized geometries, assignments and electronic structure calculations for the compound. The structure of the compound has been optimized by using the DFT (B3LYP) method with the 3-21G basis sets, using the Gaussian 09 program.Density functional theory methods were employed to determine the optimized structures of C20H11F13ClN3O3S and Initial calculations were performed at the DFT level and split- valence plus polarization 3-21G basis sets were used.
2. Experimental
2.1 Chemicals and reagents
The optimized structural parameters were used in the vibrational frequency calculations at the DFT level to characterize all stationary points as minima. All computational are carried out using Gaussian 09w program. Harmonic vibrational frequencies (ν) in cm-1 and infrared intensities (int) in Kilometer per mole of all compounds were performed at the same level on the respective fully optimized geometries. Energy minimum molecular geometries were located by minimizing energy, with respect to all geometrical coordinates without imposing any symmetrical constraints.
2.2 NBO (Natural Bond Orbital) study of structures
NBO Calculated Hybridizations are significant parameters for our investigation. The structure of the compound has been optimized by using the DFT (B3LYP) method with the 3-21G basis sets, using the Gaussian 09w program. Density functional theory methods were employed to determine the optimized structures of C20H11F13ClN3O3S (Table 1, Figure 1).
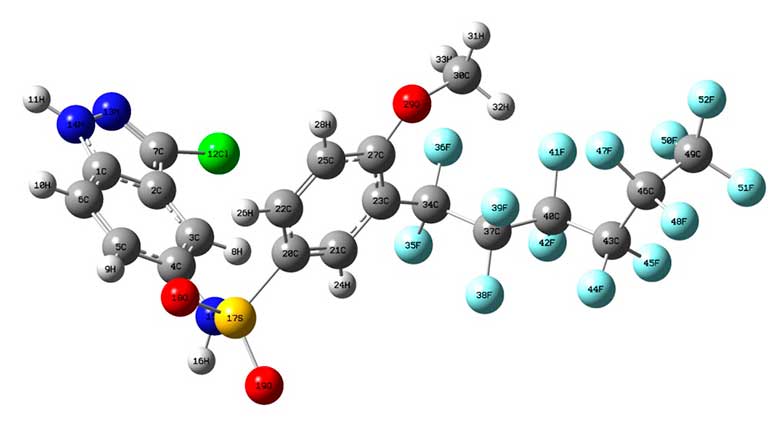
Figure 1. Optimized geometries of C20H11F13ClN3O3S at B3LYP/3-21Glevel of theory.
Table 1. Geometrical parameters optimized for C20H11F13ClN3O3S some selected bond lengths (Å) and angles (◦).
|
|
Bond length |
|
Bond length |
|
Bond length |
|
C7-Cl12 |
1.7959 |
H31-F52 |
0.9053 |
C43-F44 |
1.35 |
|
N13-N14 |
1.4114 |
H33-F39 |
1.3936 |
C43-F45 |
1.35 |
|
N15-S17 |
1.7765 |
C34-F35 |
1.35 |
C46-F47 |
1.35 |
|
S17-O18 |
105992 |
C37-F39 |
1.35 |
C46-F48 |
1.35 |
|
S17-O19 |
1.5874 |
C40-F42 |
1.35 |
C49-F50 |
1.35 |
|
O29-C30 |
1.4639 |
F41-F52 |
1.717 |
C49-F51 |
1.35 |
|
C30-F52 |
1.4049 |
F41-F50 |
1.4323 |
F41-C49 |
1.7674 |
|
|
Bond Angles |
|
Bond Angles |
|
Bond Angles |
|
N15-S17-O19 |
102.3412 |
F35-C34-F36 |
109.4712 |
H32-C46-F48 |
150.7584 |
|
C2-C7-Cl12 |
124.3832 |
F38 |
109.4712 |
F47-C46-F48 |
109.4713 |
|
O29-C30-F52 |
111.7984 |
F41-C40-F42 |
109.4712 |
F41-C49-F51 |
151.1907 |
|
Cl12-C7-N13 |
121.5988 |
C40-F41-F50 |
108.903 |
F50-C49-F52 |
109.4713 |
|
C7-N13-N14 |
103.8277 |
C40-F41-F52 |
124.9545 |
F51-C49-F52 |
109.4712 |
|
C30-H32-F47 |
134.5953 |
F50-F41-F52 |
88.3745 |
C30-F52-F41 |
73.4312 |
|
N15-S17-O18 |
114.0423 |
F44-C43-F45 |
109.4712 |
H31-F52-F41 |
51.3647 |
Natural Bond Orbital's (NBOs) are localized few-center orbital's that describe the Lewis-like molecular bo nding pattern of electron pairs in optimally compact form. More precisely, NBOs are an orthonormal set of localized "maximum occupancy" orbital's whose leading N/2 members (or N members in the open-shell case) give the most accurate possible Lewis-like description of the total N-electron density. This analysis is carried out by examining all possible interactions between "filled" (donor) Lewis-type NBOs and "empty" (acceptor) non-Lewis NBOs, and estimating their energetic importance by 2nd-order perturbation theory. Since these interactions lead to donation of occupancy from the localized NBOs of the idealized Lewis structure into the empty non-Lewis orbitals (and thus, to departures from the idealized Lewis structure description), they are referred to as "delocalization" corrections to the zeroth-order natural Lewis structure.
Natural charges have been computed using natural bond orbital (NBO) module implemented in Gaussian 09w. The. These quantities are derived from the NBO population analysis. The former provides an orbital picture that is closer to the classical Lewis structure. The NBO analysis involving hybridizations of selected bonds are calculated at B3LYP methods and 3-21G level of theory (Tables 2, 3).
Table2. The NBO Calculated Hybridizations for C20H11F13ClN3O3S at the B3LYP/3-21G.
|
B3LYP |
Atom |
Bond |
B3LYP |
Atom |
Bond |
|
S1P4.55,S1P2.73 |
N13-N14 |
N-N |
S1P2.83,S1P6.03 |
C7-Cl12 |
C-Cl |
|
S1P2.22,S1P8.76 |
S17-O18 |
S-O |
S1P4.51,S1P4.18 |
N15-S17 |
N-S |
|
S1P4.19,S1P3.18 |
C4-F35 |
C-F |
S1P2.58,S1P8.76 |
S17-O19 |
S-O |
|
S1P2.59,S1P4.76 |
O29-C30 |
O-C |
S1P2.57,S1 |
N15-H16 |
N-H |
|
S1P3.98,S1P3.12 |
C37-F38 |
C-F |
S1P4.16,S1P3.16 |
C34-F36 |
C-F |
|
S1P4.07,S1P3.23 |
C40-F41 |
C-F |
S1P4.19,S1P3.22 |
C37-F39 |
C-F |
|
S1P3.97,S1P3.19 |
C43-F44 |
C-F |
S1P4.00,S1P3.14 |
C40-F42 |
C-F |
These data shows the hyper conjugation of electrons between ligand atoms with central metal atom. These conjugations stand on the base of p-d π-bonding. The NBO calculated hybridization for C20H11F13ClN3O3S shows that all of compounds have SPX hybridization and non planar configurations. The total hybridization of these molecules are SPX that confirmed by structural. The amount of bond hybridization showed the in equality between central atoms angles (Table 2) shown distortion from normal VSEPR structures and confirmed deviation from VSEPR structures. (Figure 2).
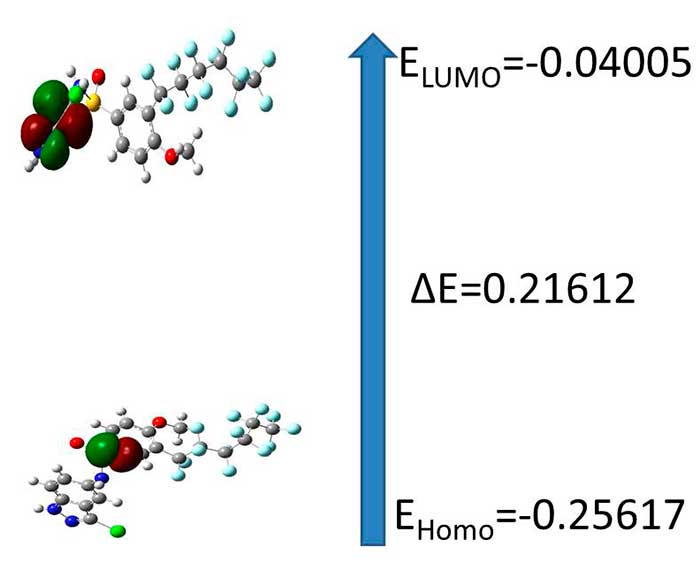
Figure 2. The atomic orbital of the frontier molecular orbital for C20H11F13ClN3O3S at B3LYP/3-21G level of theory
Some thermodynamic parameters Frequencies for C20H11F13ClN3O3S Zero-point Energy, correction Energy, Enthalpy lengths, Gibbs free Energy are calculated and confirmed with other published theoretical data ( Table 4).
Table 3. Some thermodynamic parameters Frequencies for C20H11F13ClN3O3S Zero-point Energy,correction Energy, Enthalpy lengths, Gibbs free Energy.
|
C20H11F13ClN3O3S |
|
Zero-point correction= 0.314221 (Hartree/Particle) |
|
Thermal correction to Energy= 0.351115 |
|
Thermal correction to Enthalpy= 0.352059 |
|
Thermal correction to Gibbs Free Energy= 0.240518 |
2.3 Electronic density
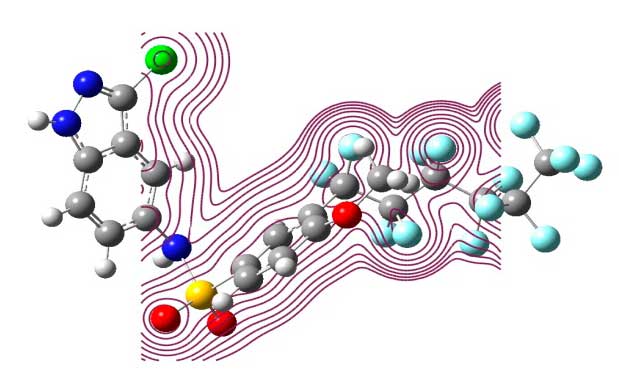
Figure 3. Total electron density surfaces for the C20H11F13ClN3O3S
2.4 Frontier molecular orbital
The HOMO represents the ability to donate an electron, LUMO as an electron acceptor represents the ability to obtain an electron. The HOMO and LUMO energy were calculated by B3LYP/3-21G method. This electronic absorption corresponds to the transition from the ground to the first excited state and is mainly described by one electron excitation from the highest occupied molecular or orbital (LUMO) both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the main orbital take part in chemical stability.. Therefore, while the energy of the HOMO is directly related to the ionization potential, LUMO energy is directly related to the electron affinity. Energy difference between HOMO and LUMO orbital is called as energy gap that is an important stability for structures. In addition, 3D plots of highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) are shown in Figure 2. The HOMO–LUMO energies were also calculated at the3-21G and the values are listed in Figure respectively.
IR spectrum
Infrared spectra may be obtained from samples in all phases (liquid, solid and gaseous). Liquids are usually examined as a thin film sandwiched between two polished salt plates (note that glass absorbs infrared radiation, whereas NaCl is transparent). If solvents are used to dissolve solids, care must be taken to avoid obscuring important spectral regions by solvent absorption. Perchlorinated solvents such as carbon tetrachloride, chloroform and tetrachloroethene are commonly used. Alternatively, solids may either be incorporated in a thin KBr disk, prepared under high pressure, or mixed with a little non-volatile liquid and ground to a paste (or mull) that is smeared between salt plates.but,in this paper we obtained IR spectrum theoretically by use Gaussian 09.frequencies of functional group showed in Figure 4.
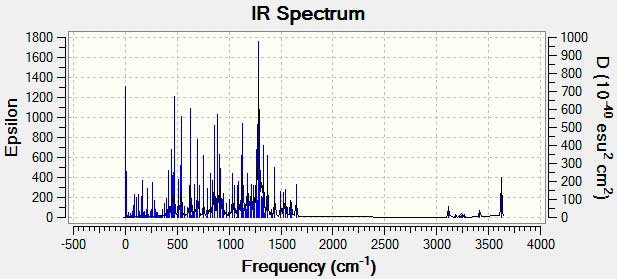
Figure 4. IR spectrum for C20H11F13ClN3O3S
NMR spectrum
Figure 5. NMR spectrum for C20H11F13ClN3O3S
Conclusion
In this research we are interested and studied on fluorocarbon compound were chosen to theoretical studies. The optimized geometries and frequencies of the stationary point and the minimum-energy paths are calculated by using the DFT (B3LYP) methods with 3-21G basis sets. B3LYP/3-21G calculation results indicated that some selected bond length and bond angles values for the C20H11F13ClN3O3S. The group point of compound is C1.
Acknowledgement
We gratefully acknowledge the financial support from the Research Council of Imam Khoemieni International University by Grant No, 751387-91 and many technical supports that provided by Tarbiat Modaress University.
References
- Ghadah S. Alghamdi and Ali. Z. Alzahrani, Bonding Formation and Orbitals Nature of Zno Structure .2013.13.9.751
- Monika, W. A.; Siddique, A. D. Portugaliae ElectrochimActa 2005, 23, 445.
- O'Hagan, D . February 2008. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 37 (2): 308–19
- Sudha. S,Sundaraganesan.N,Kurt,Cinar.M. 2011 Karabacak, Molecularstructure, vibrational spectroscopic, first order hyperpolarizability and HOMO, LUMO studies of 2-aminobenzimidazole.Journal of Molecular Structure,84: 184-195.
- Lemal, D. M. 2004. Perspective on fluorocarbon c?hemistry. Journal of Organic Chemistry, 69 (1): 1–11.
- Murphy, C.D, Schaffrath, C. O'Hagan D2003Fluorinated natural products: the biosynthesis of fluoroacetate and 4-fluorothreonine in Streptomyces cattleya" Chemosphere,52(2):455-61.
- Lewandowski ,G. Meissner E, Milchert E.Hazard ,J. 2006. Special applications of fluorinated organic compounds.136(3):385–91.
- Becke, A. D. 1993 Density-Functional Thermochemistry. III. The Role of Exact
Exchange. J. Chem. Phys., 98: 5648-5652 - Sundaraganesan, N. and S. Ilakiamani, 2007. Dominic Joshua B Vibrational spectroscopy investigation using ab initio and density functional theory analysis on the structure of 3, 4-dimethylbenzaldehyde. Spectrochimica Acta Part A., 68: 680-687
- Lewis, D. F. V., C. Ioannides, and Parke, D. V. 1994. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: computer analysis of structure- activity relationships. Xenobiotica, 24: 401-408.
- Ralph, G. 1992. Chemical hardness and the electronic chemical potential Inorganic, chimica Acta, 198: 781-786.
- Zhang, W.Curran D.P. 2006. Synthetic Application of Fluorous. Tetrahedron 62: 11837–11865.
- Smith, M. C., Ciao.Y, Wang ,H. andGeorge, S. J. 2005. Coucouvanis D., Koutmos M, Sturhahn W, Alp EA, Zhao J, Kramer SP Normal-Mode Analysis of FeCl4- and Fe2S2Cl42- via Vibrational Mossbauer, Resonance Raman, and FT-IR Spectroscopies.Inorg. Chem., 44: 5562-5570.
- Vrajmasu, V. V., Mu¨nck,E. and E. L. Bominaar, 2004. Theoretical Analysis of the Jahn−Teller Distortions in Tetrathiolato Iron(II) Complexes. Inorg. Chem., 43: 4862–4866.
- Ghammamy, Sh., K. Mehrani,Rostamzadehmansor, S. and Sahebalzamani,H. 2011. Density functional theory studies on the structure, vibrational spectra of three new tetrahalogenoferrate (III) complexes. Natural Science, 3, 683-688.
- Frisch, M. J. Trucks, G. W. 1998. GASSIAN 98 (Revision A. 3) Gaussian Inc., Pittsburgh, PA, USA.
Recommended for publication by Prof. S. Igoumnov
Fluorine Notes, 2015, 101, 3-4
