Received: November, 2014
Fluorine Notes, 2014, 97, 3-4
Synthesis of 4-fluoroalkyl-9H-pyrimido[4,5-b]indoles
Alexander S. Golubev*, Olga A. Mityushina, Alexander F. Shidlovskii, Kyrill Yu. Suponitsky, Nikolai D. Chkanikov
A.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilov St.
28, GSP-1, Moscow, Russian Federation
Fax: (499) 135 5085. E-mail: golubev@ineos.ac.ru
Abstract: Synthesis of 4-fluoroalkyl-2-(methylthio)-9H-pyrimido[4,5-b]indoles 1 starting from the easily available 6-fluoroalkyl-2-(methylthio)pyrimidin-4(3H)-ones 2 has been developed. An intramolecular Heck cyclisation of 4-(anilino)-5-iodo-2-methylthio-6-(fluoroalkyl)pyrimidines 5 was a key step of the synthesis.
Keywords: 9H-pyrimido[4,5-b]indoles, 2-methythio-4-fluoroalkyl-9H-pyrimido[4,5-b]indoles, intramolecular Heck cyclisation, 6-fluoroalkyl-2-(methylthio)pyrimidin-4(3H)-ones
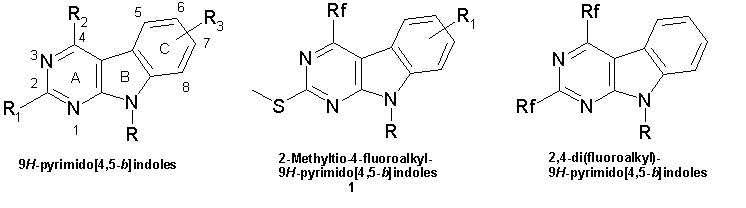
9H-Pyrimido[4,5-b]indoles are an important class of heterocycles. The first examples of this ring system were synthesized by R. G. Glushkov and O. Yu. Magidson at S. Ordzhonikidze All-Union Scientific Research Chemicopharmaceutical Institute, Moscow [1].
9H-Pyrimido[4,5-b]indoles possess important biological properties [2-7]. 9H-Pyrimido[4,5-b]indole ring system is the basic skeleton of numerous compounds possessing anticancer activities. 9H-Pyrimido[4,5-b]indoles have been recently [7] used to design new tubulin polymerization inhibitors as microtubule-targeting anticancer agents [8]. The authors have documented an importance of several pharmacophoric points in a 9H-pyrimido[4,5-b]indole-based tubulin polymerization inhibitor, namely, a rigid skeleton, presence of hydrophobic substituent at C4 of pyrimido[4,5-b]indole framework, hydrogen bond acceptors on the ring C, and hydrogen bond donors on the ring A [7].
The introduction of fluoroalkyl substituents into a newly designed inhibitor is a known method to provide binding affinity of an inhibitor with hydrophobic pockets of an enzyme. [9].
The aim of this work is the synthesis of 4-fluoroalkyl-2-(methylthio)-9H-pyrimido[4,5-b]indoles (1), which derivatives might act as an tubulin polymerization inhibitor. Known 2,4-di(fluoroalkyl)-9H-pyrimido[4,5-b]indoles were obtained via inverse electron-demand Diels–Alder (IDA) reactions of 2-aminoindoles with 2,4,6-tris(fluoroalkyl)-1,3,5-triazines [10,11]. 2,4-Di(fluoroalkyl)-9H-pyrimido[4,5-b]indoles have had significant limitations in the designing of inhibitors as substituent variation at C2 was not possible in the frame of the IDA approach. On the contrary the use of 2-alkylthio substituents at C2 is a convenient approach for the preparation of differently C2-substituted pyrimidines [12].
9H-Pyrimido[4,5-b]indoles (1) can be constructed either from the indole framework [13a,b] or from the pyrimidine ring [13c]. Pursuing the latter approach we should take into account the strong electron withdrawing effect of fluoroalkyl substituents. They deactivate the pyrimidine ring towards electrophilic attack to a great extent. This factor would hamper obtaining 9H-pyrimido[4,5-b]indoles by reactions, which are driven by electron donor ability of the pyrimidine ring, in particular, Nenitzescu reaction [14] or variants of Fisher [7,15] and Bischler [2,16] indole synthesis. But palladium-catalyzed aryl bromide or aryl iodide reactions (various cross-coupling transformations, Heck reaction) are facilitated by strong electron withdrawing substituents on an aromatic ring [17]. 5-Iodopyrimidines have been previously used in the synthesis of 4-unsubstituted 9H-pyrimido[4,5-b]indoles by an intramolecular Heck [18] and Stille cross-coupling [19]. We believed that an intramolecular Heck reaction would be a good tool for the synthesis of 4-substituted 9H-pyrimido[4,5-b]indoles, in particular, 4-fluoroalkyl-2-(methylthio)-9H-pyrimido[4,5-b]indoles (1).
Results and discussion
We report here four-step synthesis of 4-fluoroalkyl-9H-pyrimido[4,5-b]indoles (1) starting from the easily available 6-fluoroalkyl-2-(methylthio)pyrimidin-4(3H)-ones (2) (scheme 1). The synthetic procedure constitutes several successive transformations, namely, iodination of compounds (2) at C5 of the pyrimidine ring to give 5-iodopyrimidones (3), further chlorination to form 4-chloropyrimidines (4) followed by reaction of pyrimidines (4) with anilines to give 4-anilino-5-iodopyrimidines (5). The key step of our approach was an intramolecular Heck cyclisation of pyrimidines (5). Among the fluoroalkyl substituents, we took into account trifluoromethyl CF3- and difluoromethyl CF2H- groups.
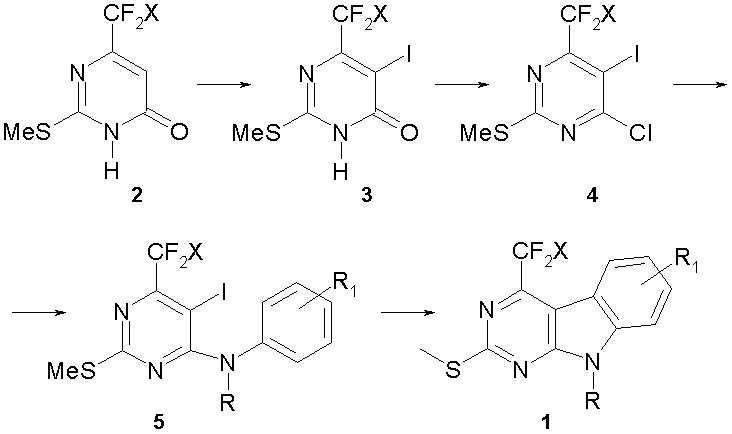
Scheme 1
6-Fluoroalkyl-2-(methylthio)pyrimidin-4(3H)-ones (2) are either commercially available (like 6-CF3-analog (2а)) or can be easily obtained by cyclocondensation of a bidentate nucleophile S-methylisothiourea hemisulfate with ethyl 4,4,4-trifluoroacetoacetate or ethyl 4,4-difluoroacetoacetate (scheme 2). The best yields of compounds (2a,b) were achieved with 10% NaOH aqueous solution [20].
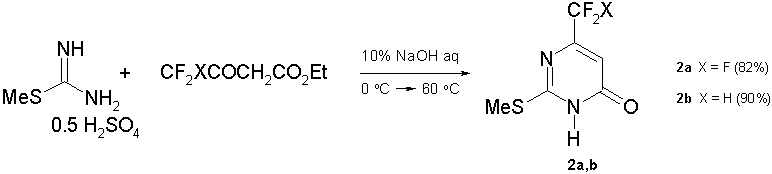
Scheme 2
The common procedure for С5-iodination of pyrimidines, namely, reaction with iodine in NaOH aqueous solution [21], led to unsatisfactory results when it has been applied to pyrimidinones (2a,b). Iodination with N-iodosuccinimide (NIS) in boiling acetonitrile [22] afforded the best results to provide 5-iodopyrimidones (3a,b) in a high yield (scheme 3).
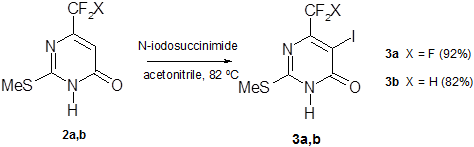
Scheme 3
A somewhat smaller yield of CF2H-analog (3b) in comparison with compound (3а) is due to purification losses of the former. Separation of pyrimidones (3a,b) from succinimide was accomplished by heating in water at 50°С. Substantial solubility of CF2H-analog (3b) in water at 50°С causes the NIS iodination yield to decrease. At the same time the more hydrophobic CF3-group induces poor water solubility of CF3-analog (3a) to prevent product losses in the purification process.
The boiling of pyrimidinones (3a,b) in phosphorus oxychloride POCl3 without a tertiary amine addition resulted in a rather low conversion of them into chloropyrimidines (4a,b). The common synthetic method for chlorination of related pyrimidinones at position 4 is the boiling in phosphorus oxychloride in the presence of N,N-dimethylaniline [23]. For pyrimidinones (3a,b), this method met with a moderate success. Chloropyrimidines (4a,b) obtained were contaminated with byproducts due to a side reaction with N,N-dimethylaniline and had to be purified by column chromatography. The best results were obtained by a combination of phosphorus oxychloride and DBU (1,8-diazabicyclo[5.4.0]undec-7-ene). With DBU (50-60 mol%), chlorination of pyrimidinones (3a,b) proceeded smoothly to give exclusively chloropyrimidines (4a,b) in a high yield (scheme 4).
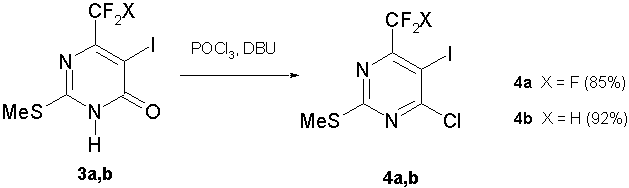
Scheme 4
Synthesis of 4-(anilino)pyrimidinones (5a-с) was accomplished by reaction of chloropyrimidines (4a,b) with the corresponding aniline (taken in excess of 15-20 mol%) in the presence of equimolar amounts of N,N-diisopropylethylamine (Huenig base) and LiBr in boiling dioxane (scheme 5).
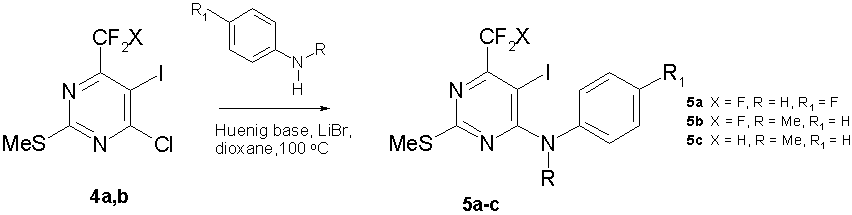
Scheme 5
LiBr as an additive caused reaction times to decrease and yields of 4-(anilino)pyrimidinones (5a-с) to increase.
An intramolecular Heck cyclisation was conducted by heating a mixture of an 4-(anilino)pyrimidinone (5) (1 equiv), Pd(OAc)2 (10 mol%), tricyclohexylphosphine PCy3 (20 mol%) and sodium acetate (2 equiv) in DMF at 125 °С [18]. In the case of compound (5a) we observed its full conversion and formation of two reaction products after 4 h. The minor product was isolated in 25% yield and turned out to be 9H-pyrimido[4,5-b]indole (1а) (scheme 6). The main product was deiodinated 4-(anilino)pyrimidinone (6а) isolated in 60% yield. All attempts to increase the yield of compound (1а) failed. So, triphenylphosphine PPh3 instead of PCy3 produced the same low yield of (1а). Triethylamine or KF as a base have not improved a ratio of products. Attempts to increase the reaction concentration up to 0.5 М met with no success. Jeffery’s “ligand-free” protocol was successfully applied for synthesis of some nitrogen-containing heterocycles by an intramolecular Heck cyclisation [24]. But, when we heated a mixture of compound (5a) (1 equiv), Pd(OAc)2 (10 mol%), tetrabutylammonium bromide (1 equiv), and potassium carbonate (3 equiv) in DMF at 125 °С, 9H-pyrimido[4,5-b]indole (1а) was not isolated at all.
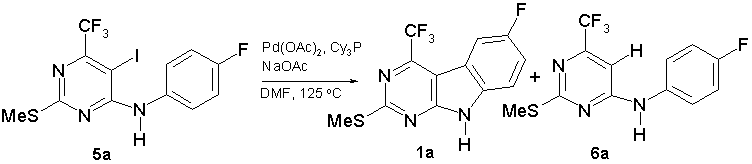
Scheme 6
Much to our delight, when the same reaction conditions: Pd(OAc)2 (10 mol%), PCy3 (20 mol%), sodium acetate (2 equiv), DMF as a solvent, 125°С, were applied for N-methylated analogs (5b,c), the target 9H-pyrimido[4,5-b]indoles (1b,c) were isolated in a satisfactory yield (scheme 7).
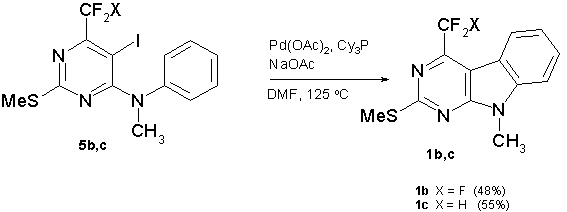
Scheme 7
The structures of 9H-pyrimido[4,5-b]indoles (1a-c) were confirmed by NMR spectroscopy, mass spectrometry, elemental analysis, and X-ray diffraction (for (1с)). The general view of structure (1с) with the atomic numbering adopted in crystallographic experiments is shown in Fig. 1.
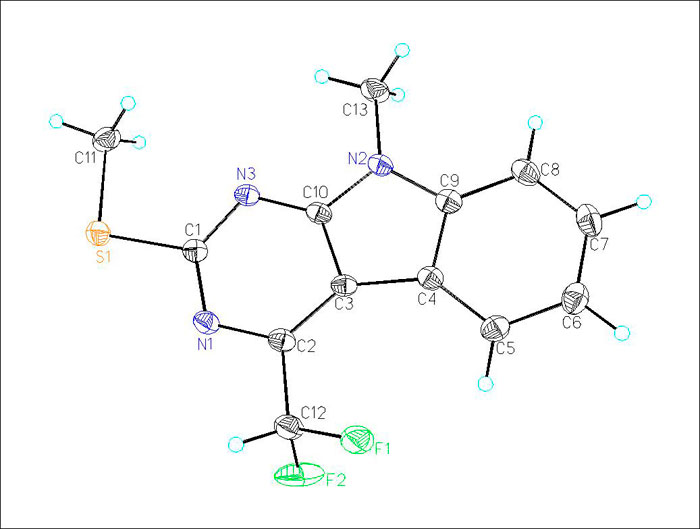
Fig.1. General view of structure (1с) with thermal displacement ellipsoids (p = 50%).
Experimental
1H NMR spectra were recorded on a Bruker AvanceTM300 spectrometer (300.13 MHz) or on a Bruker AvanceTM600 spectrometer (600.21 MHz). 13C{1H} NMR spectra were recorded on a Bruker AvanceTM600 spectrometer (150.93 MHz). 19F NMR spectra were recorded on a Bruker AvanceTM300 spectrometer (282.38 MHz). Chemical shifts for 19F nuclei were determined with respect to trifluoroacetic acid as an external standard.
Mass spectra were obtained on a Finnigan Polaris Q instrument (ion trap, 70 eV, the DIP procedure was used for the sample injection). Silica gel with the particle size of 0.06—0.20 mm (Merck Kieselgel 60) was used for column chromatography. Monitoring of the reaction course and purity of products obtained was performed using Merck Kieselgel 60 F254 TLC plates. N,N-Dimethylformamide (99.8%, Catrosa) was purified by distillation in vacuo over СаH2. Ethyl 4,4,4-trifluoroacetoacetate was purchased from «P&M», ethyl 4,4-difluoroacetoacetate was purchased from «Aldrich». Elementar analysis was performed in A.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences. Light petrolem (LP), ethyl acetate (EA), and N,N-dimethylformamide (DMF) were used as the solvents.
2-Methylthio-6-(trifluoromethyl)pyrimidin-4(3H)-one (2a).
To a solution of S-methylisothiourea hemisulfate (7.0 g, 50 mmol) in aqueous 10% NaOH (40 мl) was added ethyl 4,4,4-trifluoroacetoacetate (9.2 g, 50 mmol) dropwise at 0 oC. The reaction mixture was stirred for 2 h at 0-5 °С, allowed to warm to rt, heated at 65 °С for 1 h, cooled down to 20 °С, and poured into ice water. Water solution was acidified to рH 6 with acetic acid. A precipitate formed was filtered off, washed thoroughly with water and air-dried to give compound (2a) (8.6 g, 82%) as white crystals. M.p. 178-179 oC (see [23]: 179-180 °С). 1H NMR (600.22 MHz, CDCl3): δ 6.64 (1H, с, H5), 2.70 (3H, s, S-Me). 13С NMR (150.93 MHz, d6-DMSO): δ 166.6, 163.0, 151.5 (q, 2JC,F= 34 Hz, C6-CF3), 121.0 (q, 1JC,F= 274 Hz, CF3), 108.0, 13.4. 19F NMR (282.38 MHz, CDCl3): δ 5.85 (s, CF3). EIMS 70 eV, m/z: 211 [M + H]+ (11), 210 [M]+ (100), 191 [M – F]+ (10), 190 [M – HF]+ (56), 163 [M – SMe]+ (48), 162 [M – H–SMe]+ (24), 141 [M – CF3]+ (22).
6-Difluoromethyl-2-(methylthio)pyrimidin-4(3H)-one (2b) was obtained similarly from S-methylisothiourea hemisulfate and ethyl 4,4-difluoroacetoacetate. Yield 90%. White crystals. M.p. 201-202 oC. 1H NMR (300.13 MHz, d6-DMSO): δ 6.88 (1H, t, J = 54 Hz, CF2H), 6.35 (1H, s, H5), 2.50 (3H, s, S-Me). 13С NMR (150.93 MHz, d6-DMSO): δ 165.7, 163.5, 157.1 (t, 2JC,F= 23 Hz, C6-CF2H), 112.5 (t, 1JC,F= 237 Hz, CF2H), 107.2, 13.3. 19F NMR (282.38 MHz, d6-DMSO): δ -45.06 (s, CF2H). EIMS 70 eV, m/z: 193 [M + H]+ (12), 192 [M]+ (100), 191 [M– H]+ (56), 172 [M – HF]+ (65), 145 [M – SMe]+ (27), 144 [M – H – SMe]+ (29), 141 [M – CF2H]+ (46). Anal. Calcd for C6H6F2N2OS: C, 37.50; H, 3.15; N, 14.58. Found C, 37.33; H, 3.17; N, 14.44.
5-Iodo-2-methylthio-6-(trifluoromethyl)pyrimidin-4(3H)-one (3a).
Compound (2a) (3.15 g, 15 mmol) and N-iodosuccinimide (3.82 g, 17 mmol) were combined in acetonitrile (40 ml). The suspension obtained was refluxed for 2 h and cooled down to rt. Acetonitrile was distilled off in vacuo. The residue was dissolved in ethyl acetate (50 ml) and treated with 5% aqueous Na2S2O3 untill the organic layer was decolorized. The organic layer was separated and evaporated in vacuo. To the residue (4.8 g of beige crystalls) was added water (50 ml). The suspension obtained was heated to 50 °С for 0.5 h, cooled down to rt, filtered off, and air-dried to give compound (3a) (4.63 g, 92%) as beige crystals. Т.пл. 241-243 oC (ethanol/water). 1H NMR (300.13 MHz, d4-methanol): δ 2.65 (3H, с, S-Me). 13С NMR (150.93 MHz, d4- methanol): δ 163.8, 161.4, 154.3 (q, 2JC,F= 33 Hz, C6-CF3), 120.5 (q, 1JC,F= 276 Hz, CF3), 82.8. 12.2. 19F NMR (282.38 MHz, d4- methanol): δ 9.93 (s, CF3). EIMS 70 eV, m/z: 337 [M + H]+ (15), 336 [M]+ (84), 289 [M – SMe]+ (8), 210 [M + H – I]+ (16), 209 [M – I]+ (100). Anal. Calcd for C6H4F3IN2OS: C, 21.44; H, 1.20; N, 8.34. Found C, 21.22; H, 0.98; N, 8.21.
6-Difluoromethyl-5-iodo-2-(methylthio)pyrimidin-4(3H)-one (3b) was obtained similarly from compound (2b). Yield 82%. Beige crystals. M.p. 249-250 oC (ethanol/water). 1H NMR (300.13 MHz, d6-DMSO): δ 11.13 (1H, br.s, NH), 6.95 (1H, t, J = 53 Hz, CF2H), 2.61 (3H, s, S-Me). 1H NMR (300.13 MHz, CDCl3): δ 10.81 (1H, br.s, NH), 6.79 (1H, t, J = 53 Hz, CF2H), 2.71 (3H, s, S-Me). 13С NMR (150.93 MHz, d6-DMSO): δ 163.9, 160.8, 156.4 (t, 2JC,F= 21Hz, C6-CF2H), 114.9 (t, 1JC,F= 242Hz, CF3), 87.2. 13.5. 19F NMR (282.38 MHz, d6-DMSO): δ -42.67 (s, CF3). EIMS 70 eV, m/z: 319 [M + H]+ (9), 318 [M]+ (82), 271 [M – SMe]+ (11), 192 [M + H – I]+ (9), 191 [M – I]+ (100). Anal. Calcd for C6H5F2IN2OS: C, 22.66; H, 1.58; N, 8.81. Found C, 22.23; H, 1.43; N, 8.65.
4-Chloro-5-iodo-2-methylthio-6-(trifluoromethyl)pyrimidine (4а).
To compound (3a) (2.85 g, 8.5 mmol) was added phosphorus oxychloride (20 ml). To the yellow suspension obtained was added DBU (0.76 g, 5 mmol) dropwise at 10 °С. The reaction mixture was refluxed untill HCl evolution ceased (ca. 1.5 h). Phosphorus oxychloride was distilled off in vacuo. The residue was treated with ice water, extracted with methylene chloride (2х30 ml). The organic layer was washed with 5% aqueous NaHCO3, filtered through Celite, dried over MgSO4, and evaporated in vacuo. The residue was passed through a pad of silica gel (LP/EA 10:1) to give compound (4а) (2.55 g, 85%) as greyish crystals. M.p. 54-55 oC (LP/EA) (see [25]: 55–56 °С). 1H NMR (300.13 MHz, CDCl3): δ 2.61 (3H, s, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 173.2, 167.4, 158.6 (q, 2JC,F= 35 Hz, C6-CF3), 119.7 (q, 1JC,F= 278 Hz, CF3), 83.7, 14.6. 19F NMR (282.38 MHz, CDCl3): δ 8.75 (s, CF3). EIMS 70 eV, m/z: 355 [M + H]+ (47), 354 [M]+ (100), 334 [M – HF]+ (28). Anal. Calcd for C6H3ClF3IN2S: C, 20.33; H, 0.85; N, 7.90. Found C, 20.39; H, 0.78; N, 7.98.
4-Chloro-6-(difluoromethyl)-5-iodo-2-(methylthio)pyrimidine (4b) was obtained similarly from compound (3b) (1.91 g, 6 mmol), phosphorus oxychloride (20 ml) and DBU (0.46 g, 3 mmol). Yield 92%. Greyish crystals. M.p. 74-76 oC (LP/EA). 1H NMR (300.13 MHz, CDCl3): δ 6.70 (1H, t, J = 54 Hz, CF2H), 2.61 (3H, s, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 173.5, 166.1, 161.6 (t, 2JC,F= 24 Hz, C6-CF2H), 113.7 (t, 1JC,F= 245 Hz, CF3), 85.4, 14.6. 19F NMR (282.38 MHz, CDCl3): δ -41.60 (s, CF3). EIMS 70 eV, m/z: 337 [M + H]+ (50), 336 [M]+ (100), 316 [M – HF]+ (45). Anal. Calcd for C6H4ClF2IN2S: C, 21.41; H, 1.20; N, 8.32. Found C, 21.58; H, 1.18; N, 8.36.
N-(4-fluorophenyl)-5-iodo-2-methylthio-6-(trifluoromethyl)pyrimidin-4-amin (5а).
Compound (4a) (1.06 g, 3 mmol), 4-fluoroaniline (0.35 g, 3.15 mmol), N,N-diisopropylethylamine (0.4 g, 3.15 mmol) and LiBr (0.52 g, 6 mmol) were combined in dioxane (20 ml). The reaction mixture was refluxed for 8 h (reaction control - TLC, hexanes-EA 4:1), cooled down to rt and evaporated in vacuo. The residue was dissolved in methylene chloride (40 ml), washed with 5% aqueous HCl and brine. The organic layer was separated, dried over MgSO4, and evaporated in vacuo. The residue was purified by column chromatography on silica gel (LP/EA 4:1) to give compound (5а) (1.15 g, 90%) as greyish crystals. M.p. 97-99 oC (LP/EA). 1H NMR (300.13 MHz, CDCl3): δ 7.63 (1H, br.s, NH), 7.55 (2H, m, Ar), 7.15 (2H, m, Ar), 2.52 (3H, s, S-Me). 19F NMR (282.38 MHz, CDCl3): δ 10.43 (3F, s, CF3), -38.64 (1F, s, F-Ar). EIMS 70 eV, m/z: 430 [M + H]+ (78), 429 [M]+ (100), 428 [M – H]+ (22), 383 [M +H, – SMe]+ (12), 382 [M – SMe]+ (18), 302 [M – I]+ (19). Anal. Calcd for C12H8F4IN3S: C, 33.58; H, 1.88; N, 9.79. Found C, 33.32; H, 1.76; N, 9.80.
5-Iodo-N-methyl-2-methylthio-N-phenyl-6-(trifluoromethyl)pyrimidin-4-amine (5b) was obtained similarly from compound (4a) and N-methylaniline. Reaction time was 12 ч. Yield 78%. Greyish crystals. M.p. 130-132 oC (LP/EA). 1H NMR (300.13 MHz, CDCl3): δ 7.40 (2H, m, Ar), 7.25 (1H, m, Ar), 7.07 (2H, m, Ar), 3.57 (3H, s, N-Me), 2.61 (3H, s, S-Me). 19F NMR (282.38 MHz, CDCl3): δ 10.61 (s, CF3). EIMS 70 eV, m/z: 425 [M]+ (13), 299 [M+H – I]+ (15), 298 [M – I]+ (100), 278 [M – I– HF]+ (12). Anal. Calcd for C13H11F3IN3S: C, 36.72; H, 2.61; N, 9.88. Found C, 36.44; H, 2.20; N, 9.66.
6-Difluoromethyl-5-iodo-N-methyl-2-methylthio-N-phenylpyrimidin-4-amine (5c) was obtained similarly from compound (4b) and N-methylaniline. Reaction time was 12 ч. Yield 82%. Greyish crystals. M.p. 144-145 oC (LP/EA). 1H NMR (300.13 MHz, CDCl3): δ 7.40 (2H, m, Ar), 7.25 (1H, m, Ar), 7.09 (2H, m, Ar), 6.69 (1H, t, J = 54.5Hz, CF2H), 3.56 (3H, s, N-Me), 2.61 (3H, s, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 171.5, 163.7, 160.2 (t, 2JC,F= 25Hz, C4-CF2H), 146.0, 129.7, 126.3, 126.0, 114.6 (t, 1JC,F= 243Hz, CF2H), 73.0, 42.7, 14.4. 19F NMR (282.38 MHz, CDCl3): δ -41.33 (s, CF2H). EIMS 70 eV, m/z: 408 [M + H]+ (15), 407 [M]+ (17), 281 [M+H–I]+ (19), 298 [M – I]+ (100), 260 [M – I – HF]+ (15). Anal. Calcd for C13H12F2IN3S: C, 38.34; H 2.97; N 10.32. Found C, 38.03; H, 2.77; N, 10.01.
Experiments for obtaining 2-methylthio-4-tri(di)fluoromethyl-9H-pyrimido[4,5-b]indoles.
In a 50-ml round-bottomed flask with inert-gas inlet were combined compound (5а) (0.3 g, 0.69 mmol), Pd(OAc)2 (16 mg, 0.07 mmol), tricyclohexylphosphine (40 mg, 0.14 mmol), and sodium acetate (115 mg, 1.4 mmol). To the mixture was added dry DMF (10 ml). The suspension obtained was stirred for 0.5 h at 20 °С under argon, heated up to 125 °С, and kept for 4 h at 125 °С. The reaction mixture was cooled down to rt, poured into ice water (250 ml), and extracted with ethyl acetate (2х30 ml). The combined organic layers were washed with brine, dried over MgSO4, and evaporated. The residue was purified by column chromatography. Product of deiodination reaction N-(4-fluorophenyl)-2-methylthio-6-(trifluoromethyl)pyrimidin-4-amine (6а) was isolated first as white crystals (0.13 g, 60%). M.p. 112-113 oC (LP/EA). 1H NMR (300.13 MHz, CDCl3): δ 7.34 (2H, m, Ar), 7.14 (2H, m, Ar), 7.00 (1H, br.s, NH), 6.52 (1H, s, H5), 2.57 (3H, s, S-Me). 19F NMR (282.38 MHz, CDCl3): δ 6.91 (3F, s, CF3), -37.47 (1F, м, F-Ar). EIMS 70 eV, m/z: 304 [M + H]+ (34), 303 [M]+ (100), 302 [M – H]+ (31), 257 [M +H – SMe]+ (25), 256 [M – SMe]+ (45). Anal. Calcd for C12H9F4N3S: C, 47.52; H, 2.99; N, 13.86. Found C, 47.25; H, 2.66; N, 13.77.
Further elution of the column gave 6-fluoro-2-(methylthio)-4-trifluoromethyl-9H-pyrimido[4,5-b]indole (1а) as white crystals (0.051 g, 25%). M.p. >250oC. 1H NMR (300.13 MHz, d6-DMSO): δ 12.98 (1H, s, NH), 7.70 (1H, m, Ar), 7.62 (1H, dd, J= 7.6 Hz, J= 3.4 Hz, Ar), 7.50 (1H, td, J= 8.2 Hz, J= 1.7 Hz, Ar), 2.65 (3H, s, S-Me). 19F NMR (282.38 MHz, d6-DMSO): δ 9.54 (3F, s, CF3), -44.32 (1F, s, F-Ar). EIMS 70 eV, m/z: 302 [M + H]+ (20), 301 [M]+ (100), 300 [M – H]+ (35), 268 [M – S – H]+ (17), 255 [M + H – SMe]+ (30), 186 [M + H – SMe– CF3]+ (36). Anal. Calcd for C12H7F4N3S: C, 47.84; H, 2.34; N, 13.95. Found C, 47.64; H, 2.17; N, 13.84.
9-Methyl-2-(methylthio)-4-trifluoromethyl-9H-pyrimido[4,5-b]indole (1b) was obtained similarly from compound (5b). White crystals. Yield 48%. M.p. 143-145oC. 1H NMR (300.13 MHz, CDCl3): δ 8.26 (1H, d, J= 7.8 Hz, Ar), 7.68 (1H, t, J= 7.6 Hz, Ar), 7.54 (1H, d, J= 8.2 Hz, Ar), 7.46 (1H, t, J= 7.8 Hz, Ar), 4.00 (3H, s, N-Me), 2.79 (3H, s, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 168.1, 157.4, 146.2 (q, 2JC,F= 37.6Hz, C4-CF3), 140.4, 128.4, 123.4 (q, 5JC,F= 8.9Hz), 122.4, 121.5 (q, 1JC,F= 275Hz, CF3), 117.3, 109.6, 107.0, 28.0, 14.5. 19F NMR (282.38 MHz, CDCl3): δ 9.84 (s, CF3). EIMS 70 eV, m/z: 298 [M + H]+ (40), 297 [M]+ (100), 296 [M – H]+ (27), 264 [M – HS]+ (14), 251 [M + H – SMe]+ (42), 182 [M – SMe– CF3]+ (49). Anal. Calcd for C13H10F3N3S: C, 52.52; H, 3.39; N, 14.13. Found C, 52.49; H, 3.47; N, 13.97.
4-Difluoromethyl-9-methyl-2-(methylthio)-9H-pyrimido[4,5-b]indole (1c) was obtained similarly from compound (5c). White crystals. Yield 55%. M.p. 140-141 oC. 1H NMR (300.13 MHz, CDCl3): δ 8.26 (1H, d, J= 7.8 Hz, Ar), 7.54 (1H, t, J= 7.6Hz, Ar), 7.39 (1H, d, J= 8.2Hz, Ar), 7.36 (1H, t, J= 7.8 Hz, Ar), 6.80 (1H, t, J = 54.5Hz, CF2H), 3.85 (3H, s, N-Me), 2.71 (3H, s, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 167.6, 157.1, 151.2 (t, 2JC,F= 30Hz, C4-CF2H), 140.1, 127.8, 124.4 (t, 5JC,F= 5.5Hz), 122.1, 117.6, 115.6 (t, 1JC,F= 241Hz, CF2H), 109.4, 106.9, 27.8, 14.4. 19F NMR (282.38 MHz, CDCl3): δ -40.0 (s, CF2H). EIMS 70 eV, m/z: 280 [M + H]+ (24), 279 [M]+ (100), 278 [M – H]+ (35), 246 [M – HS]+ (20), 233 [M + H – SMe]+ (42), 213 [M – SMe– HF]+ (13), 182 [M – SMe– CF2]+ (49). Anal. Calcd for C13H11F2N3S: C, 55.90; H 3.97; N 15.04. Found C, 55.45; H, 3.88; N, 14.72.
X-Ray diffraction experiment.
Single crystals of compound (1с) suitable for X-ray study were obtained by slow evaporation of chloroform solution of compound (1с) at 20 °С. C13H11F2N3S crystals are triclinic, space group P-1: a = 8.4844(10) Å, b = 8.8967(10) Å, c = 9.4970(11) Å, α = 115.503(2)°, β =104.329(2)°, γ = 95.782(2)°, V = 609.02(12) Å3, Z = 2, M = 279.31, dcalc = 1.523 g*cm-3, μ = 0.279 mm-1. 7896 reflections were collected at SMART APEX II CCD difractometer (λ(Mo-Kα)=0.71073 Å, graphite monochromator, ω-scans, 2θ<54°) at 120K. The structure was solved by the direct methods and refined by the full-matrix least-squares procedure in anisotropic approximation. 2654 independent reflections [Rint = 0.0226] were used in the refinement procedure that has converged to wR2 = 0.0971 calculated on F2hkl (GOF = 1.051, R1 = 0.0368 calculated on Fhkl using 2304 reflections with I>2σ(I)). CCDC 1031474 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
References
- R. G. Glushkov, V.A. Volskova, O. Yu. Magidson, Pharm. Chem. J. 1967, 517-523.
- G.L. Bundy, L.S. Banitt, P.J. Dobrowolski, J.R. Palmer, T.M. Schwartz, D.C. Zimmermann, M.F. Lipton, M.A Mauragis., M. F. Veley, R. B. Appell, R. C. Clouse, E. D. Daugs, Org. Process Res. Dev., 2001, 5, 144-151.
- C.E. Mueller, U. Geis, B. Grahner, W. Lanzner, K. Eger, J. Med. Chem., 1996, 39, 2482-2491.
- H. D. H. Showalter, A. J. Bridges, H. Zhou, A. D. Sercel, A. McMichael, D. W. Fry, J. Med. Chem, 1999, 42, 5464-5474.
- P.M. Traxler, P. Furet, H. Mett, E. Buchdunger, T. Meyer, N. Lydon, J. Med. Chem., 1996, 39, 2285-2292.
- N. Zaware, H. Sharma, J. Yang, R. Kumar Vyas Devambatla, S. F. Queener, K.S. Anderson, A. Gangjee, ACS Med. Chem. Lett., 2013, 4, 12, 1148–1151
- A. Gangjee, N. Zaware, R. K. V. Devambatla, S. Raghavan, C. D. Westbrook, N. F. Dybdal-Hargreaves, E. Hamel, S. L. Mooberry, Bioorg. Med. Chem. 2013, 21, 891–902.
- (a) O. N. Zefirova, A. G. Diikov, N. V. Zyk, N. S. Zefirov, Russ. Chem. Bull., Int. Ed. 2007, 56, 4, 680-688, (b) Y. Lu, J. Chen, M. Xiao, W. Li, D. D. Miller, Pharm Res. 2012, 29, 11, 2943–2971.
- А. Hoffmann-Roeder, E. Schweizer, J. Egger, P. Seiler, U. Obst-Sander, B. Wagner, M. Kansy, D. W. Banner, F. Diederich, ChemMedChem., 2006, 1, 1205 – 1215.
- O. Iaroshenko, Synthesis 2009, 23, 3967–3974.
- G. Xu, L. Zheng, S. Wang, Q. Dang, X. Bai, Synlett, 2009, 3206-3210.
- C. L. Gibson, J. K. Huggan, A. Kennedy, L. Kiefer, J. H. Lee, C.J. Suckling, C. Clements, A. L. Harvey, W. N. Hunter, L.B. Tulloch, Org. Biomol. Chem. 2009, 7, 1829–1842.
- (а) A. S. Kumar, P. V. Amulya Rao, R. Nagarajan, Org. Biomol. Chem., 2012, 10, 5084-5093, (b) V. P. Borovik, O. P. Shkurko, Russ. Chem. Bull., Int. Ed. 2002, 51, 11, 2129-2133, (c) for synthesis of 9H-pyrimido[4,5-b]indoles from pyrimidines, see literature cited in M. Adib, B. Mohammadi, H. R. Bijanzadeh, Synlett, 2008, 177-180.
- B. Dotzauer, R. Gruenert, P. J. Bednarski, H. Lanig, J. Landwehra, R. Troschuetz, Bioorg. Med. Chem., 2006, 14, 7282–7292.
- (а) C.E. Wright, 9H-Pyrimido[4,5-b]indole-2,4-diones, J. Heterocycl. Chem., 1976, 13, 539-544; (b) C.E. Wright, J. Gambino, J. Heterocycl. Chem., 1979, 16, 401-402.
- M. A. Mauragis, M. F. Veley, M. F. Lipton, Org. Process Res. Dev., 1997, 1, 39-44.
- I. P. Beletskaya, A. V. Cheprakov, Chem. Rev., 2000, 100, 3009-3066.
- Y.-M. Zhang, T. Razler., P. F. Jackson, Tetrahedron Lett., 2002, 43, 8235-8239.
- N. Ple, A. Turck, A. Heynderickx, G. Queguiner, J. Heterocycl. Chem., 1994, 31, 1311-1315.
- Pat. JP1495267, 1964 (Chem. Abstr., 1968 , vol. 68, # 105224h).
- T. Sakamoto, Y. Kondo, R. Watanabe, H. Yamanaka, Chem. Pharm. Bull., 1986, 34, 7, 2719-2724.
- Pat. WO2010/22121 A1 2010, A. Arasappan, G.F. Njoroge, F. Bennett, V. M. Girijavallabhan, Y. Huang, R. Huelgas, J. J. Piwinski, N.-Y. Shin, V. Verma, F. Velazquez, S. Venkatraman, C. D. Kwong, S. Ananthan, J. Clark, F. Geng, H. S. Kezar, III., J. A. Maddry, R. C. Reynolds, A. Roychowdhury, J. A. Secrist, III., A.T. Fowler.
- H. Gershon, A. T. Grefig, A.A. Scala, J. Heterocycl. Chem., 1983, 20, 219-223.
- J. J. Li, J. Org. Chem. 1999, 64, 8425-8427.
- Pat. US2005/38041 A1, 2005, Y. Nakagawa, S. Bobrov, C. R. Semer IV, T. A. Kucharek,
M. Hamamoto
Recommended for publication by Prof. Nikolai D. Chkanikov
Fluorine Notes, 2014, 97, 3-4
