Received: октябрь 2012
Fluorine Notes, 2014, 96, 1-2
Химия и технология полифторированных органических соединений на основе новых агрессивостойких катализаторов
В.Ю. Захаров
Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования«Вятский
государственный университет» (ФГБОУ ВПО «ВятГУ»), 610000, г. Киров, ул. Московская 36
e-mail:
zakhar.05@mail.ru
Аннотация. Разработаны научные принципы подбора эффективных катализаторов прямого газофазного фторирования органических соединений и экспериментально показана возможность резкого увеличения селективности и скорости фторирования неразбавленным фтором при направленном изменении природы каталитической композиции. Применение созданных катализаторов позволило усовершенствовать существующие и разработать новые ресурсосберегающие, экологически более чистые технологии целого ряда полифторированных органических продуктов.
Ключевые слова: Фторорганические соединения, фтор, гетерогенный катализатор, каталитическое фторирование, изомеризация.
продолжение
2.4. Каталитическое газофазное фторирование водородсодержащих полифторированных соединений
Замещение водорода у атома углерода на фтор при прямом фторировании характеризуется, как уже отмечалось, выделением значительных количеств тепла и сопровождается, как правило, высоким вкладом деструкции субстрата. В настоящей части работы приведены результаты по исследованию прямого фторирования полифторированных водородсодержащих соединений и изучена возможность направленного качественного повышения его селективности при использовании гетерогенных контактов, созданных на основе разработанных принципов подбора эффективных катализаторов прямого фторирования.
2.4.1. Фторирование 2,5,5,9-тетрагидро-2-перфторметил-4-оксаперфторнонана.
В качестве модельного субстрата для изучения влияния природы катализатора на селективность и скорость замещения водорода в составе полифторированных соединений нами был использован 2,5,5,9-тетрагидро-2-перфторметил-4-оксаперфторнонан (ТГН). Этот субстрат содержит водород у первичного, вторичного и третичного атомов углерода; при его фторировании могут образовываться, в частности, следующие соединения:
|
Формула |
Название |
Условное обозначение |
|
CF3CF(CF3)CF2OCH2(CF2)4H |
5,5,9-тригидро-2-перфторметил-4-оксаперфторнонан |
|
|
CF3CH(CF3)CF2OCFH(CF2)4H |
2,5,9-тригидро-2-перфторметил-4-оксаперфторнонан |
|
|
CF3CF(CF3)CF2OCFH(CF2)4H |
5,9-дигидро-2-перфторметил-4-оксаперфторнонан |
|
|
CF3CF(CF3)CF2O(CF2)5H |
9-монигидро-2-префторметил-4-оксаперфторнонан |
|
|
CF3CF(CF3)CF2O(CF2)5F |
2-перфторметил-4-оксаперфторнонан |
|
|
CF3CH(CF3)CF2OCH2(CF2)4H (субстрат) |
2,5,5,9-тетрагидро-2-перфторметил-4-оксаперфторнонан |
|
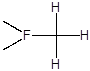
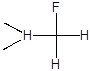
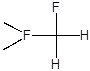
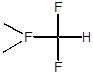

Количественный анализ состава образующихся продуктов позволяет в этом случае оценивать также относительную реакционную способность водорода в зависимости от природы связанного с ним фторуглеродного фрагмента.
Отметим, что изучение фторирования ТГН представляет практический интерес – этот процесс является одной из стадий разработанной нами совместно с ВАХЗ /185,186/ технологии утилизации перфторизобутилена (в составе продуктов пиролиза ТФЭ) с получением перспективной диэлектрической жидкости – 2-перфторметил-4-оксаперфторнонана (ПФН).
Данные по газофазному каталитическому фторированию ТГН элементарным фтором представлены в табл. 19 и 20; здесь же приведены результаты по гомогенному газофазному фторированию (в полом реакторе).
Из приведенных данных видно, что фторирование в полом реакторе без катализатора протекает неселективно с образованием в основном тетрафторметана, карбонилдифторида (фиксируется в виде CO2) и сажи (оп. 1, табл. 20) – степень деструкции сырья при этом превышает 99 % (оп. 1, табл. 19).
Фторирование в слое известных катализаторов – металлической меди (немодифицированной и покрытой серебром), и никеля, также протекает неселективно – степень деструкции сырья находится на уровне 85-95 % (оп. 2-6, табл. 19). Состав продуктов деструкции при этом несколько иной, чем при фторировании в полом реакторе – основным продуктом деструктивного фторирования является н-перфторбутан (оп. 2-6, табл. 20). Содержание ПФН в жидких продуктах фторирования невелико и находится на уровне 1-5 масс. %.
Фторирование в слое плавленого фторида кальция и альфа-оксида алюминия характеризуется некоторым увеличением селективности – степень деструкции сырья при этом уменьшается до 50-80 %, а содержание ПФН в жидких продуктах достигает 30 масс. % (оп. 7-10, табл. 19). Этот результат можно объяснить увеличением удельной поверхности контакта (по сравнению с металлическими катализаторами), что замедляет деструктивное фторирование за счет обрыва цепей этого разветвленного радикального процесса.
Выше нами было показано, что скорость и селективность прямого фторирования могут быть существенно увеличены при модифицировании α-Al2O3 фторидом никеля. Использование этого катализатора при фторировании ТГН, как показали испытания, сопровождается также резким увеличением глубины и селективности фторирования (оп. 11,12, табл. 19). Так содержание ПФН в жидких продуктах фторирования (523 К) при использовании NiF2/α-Al2O3 и немодифицированного α-Al2O3 составляет 70,3 и 31,0 масс. %, а степень деструкции сырья 53 и 62 %, соответственно. Эти результаты наглядно иллюстрируют, насколько велика роль природы гетерогенного контакта при фторировании и свидетельствуют о возможности практического использования катализатора NiF2/α-Al2O3 для получения ПФН из ТГН м
Таблица 20. Состав газообразных продуктов деструктивного прямого каталитического фторирования ТГН
|
# оп. по табл. 19 |
Состав газообразных продуктов фторирования, объемн. % |
|||||||
|
CF4 |
C2F6 |
CO2 |
C3F8 |
н-C4F10 |
i- C4F10 |
i- C4F9H |
Прочие |
|
|
1 |
87,2 |
3,1 |
5,6 |
1,7 |
0,7 |
0,4 |
>0,1 |
1,3 |
|
2 |
10,5 |
8,5 |
20,3 |
11,0 |
42,4 |
1,7 |
5,2 |
0,4 |
|
3 |
8,3 |
7,5 |
16,7 |
5,3 |
49,3 |
1,2 |
7,0 |
4,7 |
|
4 |
2,8 |
1,8 |
15,1 |
7,1 |
59,1 |
1,8 |
6,8 |
5,5 |
|
5 |
11,3 |
7,7 |
11,5 |
12,8 |
46,7 |
2,4 |
5,1 |
2,5 |
|
6 |
20,1 |
7,9 |
12,7 |
12,0 |
38,0 |
2,0 |
4,4 |
2,9 |
|
7 |
24,6 |
15,9 |
15,2 |
5,4 |
25,0 |
2,5 |
10,5 |
0,9 |
|
8 |
15,8 |
10,5 |
11,4 |
4,4 |
40,4 |
2,9 |
14,0 |
0,6 |
|
9 |
8,0 |
13,8 |
13,8 |
10,5 |
28,2 |
11,0 |
11,8 |
2,9 |
|
10 |
0,6 |
10,0 |
6,8 |
8,4 |
55,7 |
9,4 |
7,1 |
2,0 |
|
11 |
0,6 |
0,6 |
18,8 |
10,9 |
52,2 |
9,1 |
5,0 |
2,8 |
|
12 |
0,4 |
1,0 |
22,8 |
17,5 |
42,0 |
10,6 |
0,8 |
4,9 |
Изучение состава продуктов селективного фторирования показывает, что активность атомов водорода в молекуле ТГН неодинакова. Так, единственным продуктом замещения двух атомов водорода в молекуле ТГН является 5,9-дигидро-2-префторметил-4-оксаперфторнонан, что свидетельствует о том, что активность водорода у третичного атома углерода и незамещенной метиленовой группы существенно выше, чем у первичного атома углерода. Количество 9-моногидро-2-перфторметил-4-оксаперфторнонана в продуктах реакции существенно выше, чем 5-моногидро-2перфторметил-4-оксаперфторнонана, что указывает на большую активность атома водорода при частично замещенной метиленовой группе (у вторичного атома углерода) по сравнению с атомом водорода у первичного атома углерода.
В дальнейшем нами была изучена возможность увеличения глубины и селективности фторирования ТГН на катализаторе NiF2/α-Al2O3 при использовании реактора из двух секций с автономными обогревами. При этом предполагалось осуществить «мягкое» фторирование с минимальной деструкцией сырья при относительно низких температурах в первой секции реактора, а образующиеся при этом полифторированные эфиры профторировать до ПФН при повышенной температуре во второй секции. Данные приведены в табл. 21.
Таблица 21. Прямое газофазное фторирование ТГН на катализаторе NiF2/α-Al2O3 в двухсекционном реакторе (объемная скорость подачи 50 час-1, мольное отношение ТГН:фтор:азот = 1:5:3).
|
# оп. |
Температура секций, К |
Степень конверсии фтора, % |
Степень деструкции сырья, % |
Состав жидких продуктов фторирования, масс. % |
|||||
|
1-ой |
2-ой |
|
|
|
C5F12-xHx x=1÷3 |
Прочие |
|||
|
1 |
443 |
443 |
82 |
28 |
58,0 |
40,3 |
0,4 |
0,5 |
0,8 |
|
2 |
443 |
493 |
87 |
35 |
79,1 |
18,6 |
1,1 |
0,5 |
0,7 |
|
3 |
453 |
493 |
89 |
44 |
89,6 |
8,7 |
0,7 |
0,6 |
0,4 |
|
4 |
453 |
523 |
90 |
46 |
91,6 |
6,8 |
0,7 |
0,6 |
0,3 |
|
5 |
453 |
623 |
90 |
48 |
91,8 |
6,0 |
1,0 |
0,5 |
0,7 |
|
6 |
473 |
623 |
92 |
52 |
94,1 |
3,4 |
1,2 |
0,5 |
0,8 |
|
7 |
493 |
623 |
94 |
53 |
94,4 |
2,8 |
1,0 |
0,9 |
0,8 |
Из приведенных данных видно, что использование реактора с двухсекционным обогревом позволяет повысить содержание ПФН в жидких продуктах до 94 масс. % без увеличения степени деструкции сырья ( ~ 50%) – температуры первой и второй зон составляют при этом 470-490 и 620 К, соответственно.
Нами была изучена также возможность исчерпывающего каталитического фторирования полифторированных эфиров, содержащихся в выделенном ректификацией ПФН, в большом избытке фтора с целью получения чистого целевого продукта, свободного от недофторированных примесей. Данные представлены в табл. 22.
Таблица 22. Очистка ПФН от недофторированных соединений прямым газофазным фторированием на катализаторе NiF2/α-Al2O3 (объемная скорость подачи 30 час-1 , мольное отношение ПФН:фтор:азот = 3:1,5:1)1.
|
# оп. |
Температура, К |
Степень деструкции ПФН, % |
Содержание недофторированных примесей, мг/кг2 |
Состав органических продуктов, масс. % |
|
|
|
|
||||
|
Исходный продукт |
1540 |
98,8 |
1,1 |
||
|
1 |
473 |
0,2 |
18 |
> 99,9 |
<0,1 |
|
2 |
523 |
0,3 |
11 |
> 99,9 |
<0,1 |
|
3 |
593 |
0,4 |
7 |
> 99,9 |
<0,1 |
|
4 |
623 |
0,4 |
4 |
> 99,9 |
<0,1 |
|
5 |
673 |
0,6 |
2 |
> 99,9 |
<0,1 |
1 Степень конверсии фтора составляла 7-15 %.
2 В пересчете на отщепляемый
фтор-ион.
Из приведенных данных следует, что фторирование полифторированных эфиров, содержащихся в ПФН, на катализаторе NiF2/α-Al2O3 в большом избытке фтора при 470-670 К позволяет достичь тонкой очистки целевого продукта от недофторированных примесей; полифторированные эфиры при этом фторируются с образованием ПФН.
Степень очистки от недофторированных соединений (их концентрацию контролировали по содержанию отщепляемого фтор-иона) увеличивается с ростом температуры фторирования и при 670 К достигает 99,9 %; остаточное содержание отщепляемого фтор-иона в ПФН при этом составляет 2-4 мг/кг (оп. 4,5, табл. 22). Отметим для сравнения, что содержание отщепляемого фтор-иона в перфтордекалине, используемом в качестве основного компонента кровезаменителя, находится на уровне 6 мг/кг. Обращает внимание низкая степнь деструкции сырья при фторировании, которая даже при 670 К не превышает 1 %. Характерно, что диэлектрическая прочность очищенного таким образом ПФН весьма велика и превышает 80 кВ (предел измерения на приборе АИМ-90).
Проведенное исследование, таким образом, свидетельствует о возможности направленного качественного увеличения селективности и скорости каталитического фторирования и для случая водородсодержащих полифторированных соединений. Отметим, что полученные данные проверены в опытно-промышленном масштабе и легли в основу разработанной технологии ПФН, включающей прямое фторирование ТГН и очистку целевого продукта от водородсодержащих примесей прямым фторированием на NiF2/α-Al2O3.
2.4.2. фторирование α-гидро-ω-хлорперфторалканов.
Фторирование H(CF2)2Cl.
1,1,2,2-Тетрафторхлорэтан (хладон-124) и октафторциклобутан (в составе их азеотропной смеси) являются неутилизируемыми многотоннажными отходами производства ТФЭ. Фторированием 1,1,2,2-тетрафторхлорэтана может быть получен пентафторхлорэтан, который легко отделяется от октафторциклобутана ректификацией; таким образом, селективное фторирование позволило бы организовать получение (из неутилизируемых фторорганических отходов) пентафторхлорэтана и октафторциклобутана – сырья для производства ГФП.
Данные по прямому фторированию азеотропной смеси 1,1,2,2-тетрафторхлорэтана и октафторциклобутана представлены в табл. 23. Видно, что газофазное гомогенное фторирование (без катализатора) протекает неселективно с образованием тетрафторметана, хлортрифторметана, трифторметана – продуктов деструктивного фторирования 1,1,2,2-тетрафторхлорэтана и октафторциклобутана. Применение немодифицированных носителей – плавленого фторида кальция и альфа-оксида алюминия приводит к заметному увеличению выхода пентафторхлорэтана. Интересно, что модифицирование α-Al2O3 фторидом меди, который, как следует из ранее представленных данных (см. рис. 1) активирует фтор весьма слабо, не приводит к заметному улучшению каталитических свойств этого контакта. Модифицирование же α-Al2O3 фторидом никеля позволило существенно увеличить селективность фторирования (до 80-90 %); октафторциклобутан при этом не реагирует.
Фторирование Н(CF2)3Cl, H(CF2)4Cl и H(CF2)8Cl.
Фторирование относительно высококипящих (по сравнению с 1,1,2,2-тетрафторхлорэтаном) α-гидро-ω-хлорперфторалканов (также являются отходами производства ТФЭ) протекает, как показали испытания, более селективно (см. табл. 24).
Таблица 24. Прямое каталитическое фторирование Н(CF2)3Cl, H(CF2)4Cl и H(CF2)8Cl (объемная скорость подачи – 100 час-1, мольное отношение фтор: субстрат = 1,5).
|
# п/п |
Катализотор |
Температура, К |
Субстрат |
|||||
|
Н (CF2)3Cl |
H (CF2)4Cl |
H (CF2)8Cl |
||||||
|
Конверсия,% |
Выход C3F7Cl, % |
Конвер-сия,% |
Выход C4F9Cl% |
Конверсия,% |
Выход C8F17Cl, % |
|||
|
1 |
α-AI2O3 |
413 |
12 |
83,7 |
15 |
99,1 |
- |
- |
|
2 |
То же |
433 |
20 |
82,4 |
23 |
97,8 |
28 |
98,6 |
|
3 |
То же |
453 |
35 |
82,0 |
48 |
97,1 |
62 |
98,0 |
|
4 |
То же |
473 |
68 |
81,5 |
93 |
96,0 |
94 |
97,4 |
|
5 |
NiF2/α-AI2O3 |
373 |
10 |
98,8 |
14 |
99,7 |
- |
- |
|
6 |
То же |
393 |
20 |
98,0 |
24 |
98,7 |
- |
- |
|
7 |
То же |
413 |
42 |
97,7 |
48 |
98,2 |
- |
- |
|
8 |
То же |
433 |
88 |
97,3 |
90 |
97,8 |
94 |
>99,9 |
|
9 |
То же |
453 |
97 |
96,8 |
98 |
97,4 |
98 |
98,7 |
Эти данные наглядно иллюстрируют каталитическое действие нанесенного фторида никеля; действительно, степень конверсии (а значит и скорость фторирования) субстратов на катализаторе NiF2/α-AI2O3 в сопоставимых условиях существенно выше, чем на немодифицированном α-AI2O3. Отметим также, что взаимодействие Н(CF2)nCl (n=3÷8) и неразбавленного фтора на катализаторе NiF2/α-AI2O3 характеризуется практически количественным выходом соответствующих продуктов селективного фторирования.
Полученные на основе отходов производства ТФЭ хлорперфторалканы могут быть превращены (при обработке фторсульфатом хлора) в соответствующие перфторалкилфторсульфаты, и далее во фторангидриды и префторкарбоновые кислоты /187-189/; в частности, на основе хлорперфторгептана может быть синтезирована перфторэнантовая кислота – эффективный фторэмульгатор, используемый в производстве фторполимеров.
2.5. Очистка перфторуглеродных жидкостей от непредельных и водородсодержащих полифторированных соединений каталитическим газофазным фторированием.
В основу отечественной технологии получения широкого класса перфторуглеродных жидкостей, как уже отмечалось, положена обработка соответсвующих углеводородов «мягким» фторирующим агентом – трифторидом кобальта; образующиеся при этом фторуглеродные жидкости (сырец) содержат значительные количества нежелательных примесей – водородсодержащих и непредельных полифторированных соединений. В настоящей части работы приведены результаты по изучению возможности тонкой очистки сырца фторуглеродных жидкостей от полифторированных примесей при их термической обработке в газовой фазе «жестким» фторирующим агентом – элементарным фтором в слое NiF2/α-Al2O3 – эффективного катализатора прямого фторирования как водородсодержащих, так и непредельных фторорганических соединений.
2.5.1. Фторирование сырца перфтордиметилперфторциклогексана.
Перфтордиметилперфторциклогексан (ПФДМЦГ) является перспективной фторуглеродной жидкостью для нанотехнологий. Кроме того, эта жидкость является эффективным диэлектриком и используется в современной авиационной технике. В /190,191/ сформулированы требования к качеству ПФДМЦГ, которые предполагают, в частности, высокую чистоту готового продукта – содержание в нем недофторированных примесей должно быть не более 40 мг/кг (в пересчете на отщепляемый фтор-ион).
Известная технология ПФДМЦГ включает термическое фторирование диметилбензола (мета-ксилола) трифторидом кобальта, очистку сырца ПФДМЦГ от недофторированных соединений экстракцией ацетоном, ректификационное выделение ПФДМЦГ, который вновь подвергают многократной промывке ацетоном с целью его окончательной очистки от недофторированных примесей.
Основными недостатками этой технологии являются:
- низкое качество ПФДМЦГ, обусловленное неполным удалением недофторированных примесей при экстракции ацетоном – их остаточное содержание более чем в 10 раз превышает допустимую концентрацию (40 мг/кг);
- низкая производительность и повышенная трудоемкость стадии многократной экстракции ацетоном;
- относительно низкий выход целевого продукта, обусловленный образованием значительных количеств неутилизируемых недофторированных соединений;
- пожаро- и взрывоопасность, обусловленные использованием ацетона;
- загрязнение окружающей среды.
Всех этих недостатков можно избежать, осуществляя очистку сырца ПФДМЦГ селективным газофазным каталитическим фторированием молекулярным фтором; действительно, такая организация технологии позволила бы повысить чистоту целевого продукта за счет снижения остаточного содержания недофторированных примесей, утилизировать полифторированные производные диметилбензола с получением целевого продукта – ПФДМЦГ, а также отказаться от использования пожаро- и взрывоопасного ацетона, и тем самым, полностью ликвидировать его сброс.
Данные по каталитическому фторированию сырца ПФДМЦГ в лабораторном и опытно-промышленном масштабе приведены в табл. 25; здесь же, для сравнения, представлены результаты, полученные при фторировании сырца в полом реакторе, без катализатора.
Из представленных данных видно, что фторирование сырца ПФДМЦГ разбавленным фтором в полом реакторе, без катализатора при 570-620 К характеризуется низкой степенью очистки от недофторированных соединений, которая находится на уровне 40-50% (оп. 1,2 табл. 25); процесс при этом протекает неселективно, с образованием значительных количеств продуктов деструкции и сажи. Сырец после обработки фтором в полом реакторе имеет характерный желтый цвет. При температурах выше 620 К происходит резкое увеличение вклада деструктивного фторирования. Использование неразбавленного фтора при фторировании в полом реакторе затруднительно, так как процесс в этом случае приобретает взрывной характер во всем исследованном интервале температур.
Таблица 25. Прямое фторирование сырца ПФДМЦГ1 (объемная скорость подачи – 140 час-1)
|
# оп. |
Катализатор |
Темпера-тура, К |
Степень конвер-сии фтора, % |
Степень деструк-ции субстра-та, % |
Содерж. нефторир примесей мг/кг |
Степень очистки от недофторир. примесей, % |
|
Результаты лабораторных опытов (мольное отношение ПФДМЦГ:фтор = 5,3) |
||||||
|
1 |
Без к-ра |
573 |
17 |
4,2 |
6020 |
52,8 |
|
2 |
То же |
623 |
51 |
5,3 |
7580 |
40,6 |
|
3 |
Cu, металл |
573 |
26 |
0,8 |
3700 |
71,0 |
|
4 |
То же |
623 |
42 |
2,4 |
4800 |
62,4 |
|
5 |
α-Al2O3 |
573 |
16 |
0,2 |
98 |
99,2 |
|
6 |
То же |
623 |
21 |
0,2 |
62 |
99,5 |
|
7 |
То же |
673 |
22 |
0,4 |
37 |
99,7 |
|
8 |
То же |
723 |
24 |
0,5 |
24 |
99,8 |
|
9 |
NiF2/α-Al2O3 |
573 |
24 |
0,1 |
50 |
99,6 |
|
10 |
То же |
623 |
28 |
0,1 |
34 |
99,7 |
|
11 |
То же |
673 |
32 |
0,2 |
18 |
99,9 |
|
12 |
То же |
723 |
35 |
0,4 |
12 |
>99,9 |
|
Результаты опытно-промышленных испытаний (мольное отношение ПФДМЦГ:фтор = 1,4) |
||||||
|
13 |
NiF2/α-Al2O3 |
573 |
- |
0,1 |
70 |
99,2 |
|
14 |
То же |
573 |
- |
0,2 |
73 |
99,2 |
|
15 |
То же |
623 |
- |
0,2 |
47 |
99,5 |
|
16 |
То же |
623 |
- |
0,2 |
48 |
99,5 |
|
17 |
То же |
673 |
- |
0,3 |
15 |
99,8 |
|
18 |
То же |
673 |
- |
0,2 |
13 |
99,9 |
1 Исходный сырец ПФДМЦГ содержал недофторированные соединения в количестве (в персчете на отщепляемый фтор-ион) 12770 мг/кг (оп. 1-12) и 9130 мг/кг (оп. 13-18). В опытах без катализатора фтор разбавляли азотом (до 25 объемн. %), в остальных опытах использовали неразбавленный фтор.
Использование известного катализатора – насадки из металлической меди, позволяет применить неразбавленный фтор; степень очистки от недофторированных примесей при этом хотя и не превышает 75 %, несколько увеличивается по сравнению с фторированием в полом реакторе (оп. 3,4, табл. 25). При температурах выше 620 К фторирование в слое металлической меди характеризуется резким увеличением степени деструкции сырья и уменьшением полноты очистки от полифторированных примесей.
Изучение фторирования сырца ПФДМЦГ на α-Al2O3 и NiF2/α-Al2O3 выявило существенное увеличение степени очистки (оп. 5-12, табл. 25), которая заметно растет с повышением температуры (до 99,9 % и более). Фторирование даже в весьма жестких условиях (720 К, неразбавленный фтор) протекает весьма селективно – так, степень деструкции сырья на NiF2/α-Al2O3 не превышает 0,4 %.
Сравнение данных, полученных с использованием α-Al2O3 и NiF2/α-Al2O3 (оп.5-12, табл. 25) свидетельствует о заметном каталитическом действии диспергированного фторида никеля – остаточной содержание недофторированных примесей на этом катализаторе может быть снижено до 10-20 мг/кг; степень очистки при этом превышает 99,9 %.
Данные по прямому фторированию сырца ПФДМЦГ в слое NiF2/α-Al2O3 на опытно-промышленной установке (оп. 13-18, табл. 25) свидетельствует о высокой эффективности каталитического способа очистки и хорошей воспроизводимости результатов при укрупнении масштабов фторирования.
Отметим, что приведенные результаты положены в основу разработанной нами и внедренной в производство технологии ПФДМЦГ повышенной чистоты, которая включает термическую обработку диметилбензола трифторидом кобальта, прямое каталитическое фторирование образующегося сырца на NiF2/α-Al2O3 и ректификационное выделение целевого продукта /192/; выпускаемый по этой технологии ПФДМЦГ, удовлетворяет предъявляемым требованиям по всем показателям (ТУ-044-13-85).
2.5.2. Фторирование сырца перфторуглеродных жидкостей и смазок.
Технология перфторуглеродных жидкостей Б-1, М-1, смазок УПИ, КС и КСТ (ПФУЖ и С), перспективных жидкостей для нанотехнологий, также как и технология ПФДМЦГ, включает термическое фторирование соответствующих углеводородов (в данном случае – фракции трансформаторного масла) трифторидом кобальта, очистку образующегося сырца от недофторированных соединений многократной экстракцией ацетоном и его фракционную разгонку при пониженном давлении с выделением целевых продуктов. Полученные таким образом ПФУЖ и С обладают, в частности, низкой хемостойкостью, обусловленной содержанием значительных количеств недофторированных примесей – к примеру, полностью разрушаются даже при кратковременном контакте с 10 %-ным водным раствором гидроксида натрия (350 К).
Повышения качества фторуглеродных жидкостей можно достигнуть, как уже отмечалось выше, при их очистке методом каталитического фторирования (взамен экстракции ацетоном); такая организация технологии позволила бы отказаться от использования пожаро- и взрывоопасного ацетона, а также увеличить выход целевых продуктов за счет утилизации полифторированных (так называемых, «ацетонорастворимых») примесей, содержание которых в сырце весьма велико и составляет 12-15 масс. %.
Данные по прямому газофазному фторированию сырца ПФУЖ и С представлены в табл. 26.
Осуществление процесса в полом реакторе (без катализатора) сопровождается неселективным деструктивным фторированием даже при разбавлении фтора азотом. Фторирование в присутствии металлического медного катализатора также протекает неселективно (оп. 1-3, табл. 26).
Использование немодифицированного α-Al2O3 позволяет достичь тонкой очистки сырца от недофторированых примесей (степень очистки достигает 99,2 %); в то же время в ряде случаев (см. напр. оп. 5, табл. 26) при использовании этого контакта наблюдалось резкое уменьшение селективности процесса.
Использование в качестве катализатора фторида никеля, нанесенного на α-Al2O3, позволило существенно увеличить степень очистки сырца ПФУЖ и С от недофторированных примесей (до 99,9 % и выше), а также стабилизировать селективное фторирование неразбавленным фтором. Таким образом, и в данном случае использование NiF2/α-Al2O3 позволяет решить проблему селективного фторирования.
Таблица 26. Прямое фторирование сырца ПФУЖ и С1 (температура – 673 К, объем реактора – 50 мл, скорость подачи сырца – 50 г/час).
|
# оп. |
Катализатор |
Скорость подачи фтора, г/час |
Степень конвер-сии фтора, % |
Степень деструкции сырья, % |
Содерж. недофто-рир. примесей мг/кг |
Степень очистки от недофто-рир. примесей % |
|
1 |
Без к-ра |
3,3 |
>99 |
24 |
15600 |
- |
|
2 |
То же |
4,4 |
>99 |
22 |
15000 |
- |
|
3 |
Cu, металл |
3,7 |
>99 |
17 |
7100 |
25,3 |
|
4 |
α-Al2O3 |
3,5 |
71 |
6 |
75 |
99,2 |
|
5 |
То же |
4,4 |
>99 |
14 |
3700 |
61,0 |
|
6 |
NiF2/α-Al2O3 |
3,4 |
73 |
4 |
54 |
99,4 |
|
7 |
То же |
4,0 |
68 |
3 |
6 |
>99,9 |
|
8 |
То же |
4,0 |
64 |
3 |
5 |
>99,9 |
1 Исходный сырец ПФУЖ и С содержал недофторированные соединения в количестве 9500 мг/кг (в пересчете на отщепляемый фтор-ион). В оп. 2 в реактор подавали азот со скоростью 6 л/час; в остальных опытах использовали неразбавленный фтор.
Интересно отметить, что осуществление очистки сырца ПФУЖ и С каталитическим фторированием позволяет более чем на два порядка понизить остаточное содержание недофторированных примесей по сравнению с методом экстракции ацетоном. Этот факт свидетельствует о существенном улучшении качества целевых продуктов, что проявляется, в частности, в повышении их хемостойкости; так, сырец после каталитической обработки фтором выдерживает даже длительный нагрев (375 К) с 40 %-ным раствором гидроксида натрия. Улучшение качества перфторуглеродных жидкостей и смазок может привести к увеличению срока их эксплуатации, а также открыть новые области для их применения.
Проведенные испытания на промышленном реакторе (количество катализатора NiF2/α-Al2O3 - 6,2л) полностью подтвердили результаты лабораторных опытов.
Широкие возможности открывает метод каталитического фторирования и для резкого (в 2 раза) увеличения выпуска дефицитных ПФУЖ и С в их существующем производстве. Увеличение производительности может быть достигнуто, как показали опытно-промышленные испытания, за счет тривиального повышения скорости подачи углеводородов при фторировании трифторидом кобальта на существующих агрегатах непрерывного действия, с последующей утилизацией интенсивно образующихся в этом случае недофторированных соединений (35-38 масс. %) селективным каталитическим фторированием элементарным фтором /193/.
Таким образом, использование селективного газофазного фторирования при очистке сырца ПФУЖ и С от недофторированных соединений (взамен экстракции ацетоном) позволяет:
- существенно улучшить качество целевых продуктов за счет резкого увеличения полноты их очистки;
- увеличить выход целевых продуктов за счет утилизации недофторированных («ацетонорастворимых») соединений, содержащихся в сырце.
- без капитальных затрат резко увеличить мощность существующего производства;
- решить проблему регенерации отработанных продуктов;
- увеличить пожаробезопасность производства и уменьшить загрязнение окружающей среды за счет полного исключения ацетона из технологии.
2.5.3. Фторирование сырца перфтордекалина.
Технология перфтордекалина (ПФД) – перспективной фторуглеродной жидкости для нанотехнологий и основного компонента искуственного кровезаменителя, включает термическое фторирование трифторидом кобальта, конденсацию сырца ПФД, его очистку от недофторированных соединений газофазным гомогенным, фторированием разбавленным элементарным фтором, тонкую очистку от неполностью фторированных примесей обработкой водно-спиртовым раствором гидроксида натрия и ректификацию /194/.
Одним из недостатков этой технологии является относительно невысокая степень очистки сырца ПФД от недофторированных соединений при его газофазном фторировании разбавленным фтором и низкая производительность узла фторирования. В этой связи представлялось интересным изучение каталитического фторирования сырца ПФД; данные представлены в табл. 27.
Таблица 27. Прямое фторирование сырца ПФД1 (температура – 673 К)
|
# оп. |
Катализатор |
Объемная скорость подачи2, час-1 |
Мольное отношение ПФД:фтор |
Степень деструкции ПФД, % |
Содерж. недоф-торир. приме-сей, мг/кг |
Степень очистки от недофто-рир. примесей, % |
|
|
1. Результаты лабораторных опытов |
|||||||
|
1 |
Без к-ра |
65 |
2,1 |
0,2 |
1312 |
94,2 |
|
|
2 |
То же |
95 |
2,1 |
1,9 |
1628 |
92,8 |
|
|
3 |
α-AI2O3 |
130 |
1,9 |
<0,1 |
241 |
98,9 |
|
|
4 |
NiF2/α-Al2O3 |
100 |
7,2 |
<0,1 |
15 |
99,9 |
|
|
5 |
То же |
110 |
3,3 |
<0,1 |
5 |
>99,9 |
|
|
2. Результаты опытно-промышленых испытаний |
|||||||
|
6 |
Без к-ра |
40 |
1,2 |
0,8 |
91 |
99,0 |
|
|
7 |
То же |
65 |
1,2 |
1,4 |
310 |
96,7 |
|
|
8 |
-«- |
65 |
1,2 |
горение (сажа, продукт желтый) |
|||
|
9 |
NiF2/α-Al2O3 |
65 |
1,2 |
0,3 |
6 |
>99,9 |
|
|
10 |
То же |
105 |
1,2 |
0,4 |
8 |
99,9 |
|
|
11 |
-«- |
130 |
1,5 |
0,4 |
17 |
99,8 |
|
|
12 |
-«- |
200 |
1,4 |
0,4 |
34 |
99,6 |
|
1 Исходный сырец ПФД содержал недофторированные соединения в количестве (в пересчете на отщепляемый фтор-ион) 22670 мг/кг (оп. 1-5) и 9450 мг/кг (оп. 6-12). В опытах без катализатора фтор разбавляли азотом до 15 объемн. % (оп. 1,2,6,7) и до 30 объемн. % (оп. 8); в остальных опытах использовали неразбавленный фтор.
2 Без учета подачи азота (подавался в опытах без катализатора).
Характерно, что гомогенное селективное фторирование (без катализатора) возможно только при разбавлении фтора азотом – в противном случае фторирование сопровождается деструкцией сырья и резким уменьшением степени очистки.
Особенностью фторирования в слое α-AI2O3 и фторида никеля, нанесенного на α-AI2O3, является возможность использования неразбавленного фтора – процесс при этом протекает селективно и характеризуется резким уменьшением остаточного содержания недофторированных соединений – примерно на два порядка по сравнению с гомогенным фторированием (оп. 1-5, табл. 27). Столь резкое увеличение степени очистки является, по-видимому, следствием как активации фтора (в случае NiF2/α-AI2O3), так и роста концентраций реагирующих компонентов (фтора и недофторированых примесей) и времени контакта при исключении подачи азота в зону реакции.
Опытно-промышленные испытания полностью подтвердили результаты лабораторных исследований (оп. 6-12, табл. 27) и свидетельствуют о существенном увеличении степени очистки и производительности реактора при фторировании неразбавленным фтором в слое катализатора NiF2/α-Al2O3. Способ каталитического фторирования сырца ПФД внедрен в его производстве.
Приведённые в настоящей работе результаты полностью согласуются с данными /195-209/ и позволяют наметить широкий круг перспективных исследований в области прямого фторирования.
Таким образом, проведенное комплексное исследование фторирования элементарным фтором широкого класса непредельных и водородсодержащих полифторированных органических соединений позволило создать научные основы подбора катализаторов для селективного их осуществления и подобрать универсальную каталитическую композицию – NiF2/α-AI2O3, обладающую высокими активностью, селективностью и стабильностью.
3. Катализ термических превращений полифторированных органических соединений. Агрессивостойкие катализаторы на основе α-AI2O3
3.1 Новые перспективы создания агреессивостойких катализаторов термических превращений полифторированных органических соединений
Особенностью термических превращений фторорганических соединений, является, как уже отмечалось, высокая реакционная активность среды, обусловленная образованием агрессивных продуктов (фтористый водород, хлористый водород, карбонилдифторид и т.д.). Это обстоятельство необходимо учитывать при разработке катализаторов термических превращений полифторированных соединений (ФОС), которые должны быть устойчивы к действию исходных веществ и продуктов реакции даже при повышенных температурах; химическое взаимодействие катализаторов с компонентами реакционных смесей сопровождается, как правило, фазовыми переходами, которые приводят к резкому снижению механической прочности и разрушению гетерогенных контактов.
Наглядным примером такого взаимодействия является глубокое фторирование γ-Al2O3 при использовании катализаторов на его основе в прямом фторировании органических соединений; соответствующий фазовый переход, как отмечалось выше, приводит к механическому разрушению этих катализаторов.
Анализ работ по созданию эффективных катализаторов термических превращений показывает, что основным приемом, обеспечивающим стабильную прочность гетерогенных контактов, является применение композиций, химически инертных по отношению к компонентам реакционных смесей в условиях данного конкретного процесса. Природа противоиона у активных центров катализатора, которыми, как правило, являются катионы металлов, определяется при этом природой реагентов. Так для интенсификации окисления кислородом наиболее часто используют оксиды металлов /210/, для осуществления превращений сераорганических соединений – сульфиды /211/, для активации молекулярного азота и водорода – нитриды и гидриды /212/, для катализа термических превращения хлор- и фторорганических соединений – хлориды и фториды металлов, соответственно /213,214/.
Катализаторы, созданные без учета этих особенностей, обладают пониженной стабильностью и, как правило, имеют ограниченный, узкий температурный интервал эксплуатации. Так, попытки промышленного использования металлического никеля, меди или хрома для окисления углеводородов или аммиака оказались неудачными, в частности, из-за окисления металлов и их постепенного разрушения /215/.
В /73/ отмечается, что температура прямого фторирования органических соединений на металлических катализаторах не должны превышать 600 К; в противном случае протекает глубокое фторирование металлов и их разрушение.
Значительно реже стабильная прочность катализаторов обеспечивается их специфической устойчивостью по отношению к компонентам реакционных смесей; так, наиболее стабильным катализатором высокотемпературного (1100-1200К) селективного окисления аммиака кислородом является металлическая платина, которая не образует оксидов и не разрушается при эксплуатации /216/.
Специфической особенностью α-Al2O3, как было показано нами, является его чрезвычайно высокая устойчивость во фторирующих средах (не разрушается в элементарном фторе даже при 920 К). Использование стабильных катализаторов на основе α-Al2O3 позволило, как показано выше, резко повысить (по сравнению с известными катализаторами) селективность и скорость прямого фторирования полифторированных органических соединений. Кроме того, α-Al2O3 весьма доступен, дешев и широко выпускается (в различных модификациях) отечественной промышленностью /217/. Все это открывает новые перспективы создания на основе α-Аl2O3 эффективных агрессивостойких катализаторов термических превращений полифторированных органических соединений.
В настоящем разделе приведены результаты исследований по катализу термического дехлорирования фторхлоруглеродов водородом и окисления тетрафторэтилена.
3.2 Дехлорирование фторхлоруглеродов водородом
Дехлорирование фторуглеродов является одним из основных способов синтеза фторолефинов – сырья для получения фторопластов, фторкаучуков и целого ряда других практически важных фторорганических продуктов. Технология дехлорирования фторхлоруглеводородов основана в настоящее время на использовании дорогого, а также дефицитного металлического цинка и характеризуется низкой производительностью, высокой металлоемкостью и загрязнением окружающей среды токсическим хлоридом цинка. Всех этих недостатков можно избежать, используя в качестве дехлорирующего агента молекулярный водород.
В тоже время для дехлорирования фторхлоруглеродов водородом даже в присутствии катализаторов необходима относительно высокая температура (600 К и выше), что приводит к протеканию побочных реакций. В этой связи актуальной является разработка активных и стабильных каталитических систем, обеспечивающих селективное дехлорирование фторхлоруглеродов водородом.
3.2.1 Аналитический обзор. Получение фторолефинов каталитическим дехлорированием фторхлоуглеродов водородом
Наиболее полно описано каталитическое дехлорирование водородом 1,1,2-трифтортрихлорэтана (хладона-113). Единственный пример осуществления этого процесса в отсутствии катализатора приведен в /218/, где в качестве реактора использовалась обогреваемая железная трубка. При 820-870 К и времени контакта 1-2 сек. степень конверсии хладона-113 составила 24-36%, а выход трифторхлорэтилена – 60-84%. В качестве побочных продуктов образуются трифтордихлорэтан (хладон-123) и трифторэтилен.
Впервые получение фторхлоруглеродов каталитическим дехлорированием насыщенных фторхлоруглеродов водородом описано авторами /219/; в качестве катализатора использовалась медь (5-10 масс.%), нанесенная на уголь или силикагель, а в качестве субстратов – 1,2-дихлортетрафторэтан, 1,1,2-трифтортрихлорэтан, 1,2-дифтортетрахлорэтан, 1,1-дифтортетрахлорэтан и 1,2-дихлогексафторциклобутан. Степень конверсии 1,1,2-трифтортрихлорэтана и выход трифторхлорэтилена на катализаторе Cu/SiO2 при 730-750 К и времени контакта 10 сек. составили 50% и 74-79%, соответственно.
Отмечается также, что металлическая медь менее активна, чем нанесенная; для достижения сравнимых степеней конверсии здесь требовалось повышение температуры до 870 К.
Дехлорирование 1,2-дифтортетрахлорэтана протекает в более мягких условиях (640-660 К); выход 1,2-дифторэтилена составляет при этом 90% на прореагировавший (72%) субстрат /219/. Степень конверсии несимметричного изомера – 1,1-дифтортетрахлорэтана в тех же условиях существенно ниже (всего 20%).
Применение в качестве катализатора дехлорирования 1,1,2-трифтортрихлорэтана никеля, кобальта, платины и палладия, нанесенных на активированный уголь, описано в /220-222/. Максимальный выход трифторхлорэтилена (75%) был достигнут на никельсодержащем катализаторе при 700-750 К, времени контакта 2,5 сек. и мольном отношении водород:хладон-113, равном 1:3.
Отмечается /223/, что использование в качестве каталитических носителей фосфатов, хлоридов и фторидов кальция и бария не приводит к повышению (по сравнению с активированным углем) эффективности катализаторов. Так, при использовании этих носителей, модифицированных никелем, выход трифторхлорэтилена составил 20-40% при 670-750 К и времени контакта 4,5 сек. Отмечается также снижение активности этих катализаторов: быстрое (в течение 2-3 часов) – вследствие образования фторидов и хлоридов никеля, и медленное (в течение 30 часов) – в результате образования на поверхности продуктов уплотнения.
В /224/ описан широкий класс катализаторов (активные компоненты – медь, кобальт, серебро или платина, носители – активный оксид алюминия, активированный уголь, стеклянные бусы или фторид магния) дехлорирования 1,1,2–трифтортрихлорэтана водородом. Максимальный выход трифторхлорэтилена – 96,7% при степени конверсии хладона-113 65% был достигнут на фториде магния, модифицированном медью и кобальтом (мольное соотношение компонентов Co:Cu:MgF2= 1:30:150) при 734 К, времени контакта 2-3 сек. и отношении хладон:водород, равном 1. Использование в качестве носителя оксида магния приводит к некоторому уменьшению селективности (до 93%); степень конверсии сырья при этом не меняется. Отмечается также /224/, что дехлорирование фторхлоруглеродов при температуре выше 870 К нежелательно вследствие снижения селективности и уменьшения пробега катализатора; катализатор может быть регенерирован кислородсодержащим газом, например, воздухом при 580-950 К.
В /225/ описан способ получения трифторхлорэтилена дехлорированием 1,1,2-трифтортрихлорэтана на фториде кальция, модифицированном оксидом хрома, а также медью или никелем. При 620-670 К и времени контакта 10 сек. степень конверсии субстрата на катализаторе Cu,Cr2O3/CaF2 составила 35-40%; практически единственным продуктом реакции при этом был трифторхлорэтилен. Оптимальное массовое соотношение Cu:Cr2O3:CaF2 при приготовлении катализатора из нитратов составляет 30:15:55. Отмечается /225/, что катализатор, содержащий только оксид хрома и фторид кальция, в этих условиях неактивен.
В то же время оксид хрома, нанесенный на стружку из алунда или плавленый оксид алюминия, может быть использован, как следует из /226,227/, в качестве катализатора дехлорирования 1,1,2-трифтортрихлорэтана; конверсия сырья и выход трифторхлорэтилена при 798 К и времени контакта 15 сек. составили 24,5 и 73%, соответственно. Отмечается повышенное время работы катализатора до регенерации (от 52 до 200 часов). В случае фторида магния, модифицированного оксидом хрома, процесс проводят при 750-820 К, времени контакта 2-15 сек. и в избытке водорода (25% от стехиометрического количества); конверсия сырья (35-40%) и выход трифторхлорэтилена (50-55%) не снижаются в течение 30 часов непрерывной эксплуатации катализатора /226/.
В /228/ описано дехлорирование 1,1,2-трифтортрихлорэтана на оксиде хрома, модифицированном медью и оксидом бария. Катализаторы готовят соосаждением гидроксидов из растворов нитратов с различным массовым соотношение металлов Ba:Cr:Cu – от 1:1,5:1,5 до 1:8:9. Оптимальным является соотношение Ba:Cr:Cu, равное 1:3:3,1; выход трифторхлорэтилена и степень конверсии сырья на таком катализаторе составили 85 и 60 %, соответственно (температура 648 К, время контакта 9 сек, мольное отношение водород:хладон-113=1,2). Отмечается /228/, что проведение реакции при температуре выше 700 К сопровождается переходом оксида хрома в каталитически неактивную форму.
В /229/ приведены данные по дехлорированию 1,1,2-трифтортрихлорэтана на активированных углях, фториде магния, шамоте и огнеупорном кирпиче, модифицированных медью, никелем, кобальтом и железом. Максимальный выход трифторхлорэтилена – 60-75 % при степени конверсии хладона-113 54-83% был достигнут на железосодержащих катализаторах (температура 770-800 К, время контакта – 4-10 сек).
В /230/ приведены результаты по дехлорированию 1,1,2-трифтортрихлорэтана водородом на натрий-магний-фториде (NaMgF3) – выход трифторхлорэтилена и степень конверсии 1,1,2-трифтортрихлорэтана при 773 К составили 83,9 и 21,3 %, соответственно. Модифицирование натриймагнийфторида никелем, медью и железом приводит, как отмечается в /230/, к существенному увеличению каталитической активности и позволяет снизить температуру процесса; так, при дехлорировании в слое медьсодержащего контакта (6,3 масс.% меди, 748 К) степень конверсии 1,1,2-трифтортрихлорэтана составила 35,6%, а выход трифторхлорэтилена – 85,2%.
Модифицирование натриймагнийфторида палладием (0,5 масс.%) приводит резкому увеличению активности катализатора /230/, что позволяет снизить температуру до 470 К; при этом происходит существенное изменение селективности дехлорирования – основным продуктом реакции является трифторэтилен. В /230,231/ также указывается на высокую каталитическую активность металлического палладия при 470-570 К в дехлорировании трифторхлорэтилена водородом до трифторэтилена.
В /232/ приведено подробное описание рецептуры и методики приготовления оксидного алюмомолибденового катализатора и отмечается возможность его использования для дехлорирования хладона-113 водородом с получением трифторхлорэтилена.
Обзор работ по дехлорированию водородом 1,2-дихлортетрафторэтана (хладона-114) показывает, что здесь используются те же типы катализаторов, что и при дехлорировании хладона-113. Так, в /233,234/ описано использование смеси оксидов меди, бария и хрома; катализатор готовят соосаждением гидроксидов меди и бария с хроматом аммония, образующийся комплекс прокаливают при 623 К и обрабатывают водородом при температуре не выше 698 К. Степень конверсии 1,2-дихлортетрафторэтана и выход тетрафторэтилена на таких катализаторах в оптимальных условиях (650-720 К, время контакта 5-10 сек, мольное отношение хладон-водород – 0,75-1,25) составляют 20-25 и 60-65%, соответственно. Отмечается, что в качестве носителя для оксидных медно-хромовых катализаторов можно использовать фториды щелочноземельных металлов, оксиды магния, алюминия и кремния.
В /235/ приведены данные по дегалоидированию водородом фторгалоидуглеродов, содержащих 2-8 атомов углерода, на фториде алюминия, модифицированном медью, родием, платиной и оксидом хрома. Для ряда катализаторов в качестве носителей использовались цеолиты и активный оксид алюминия. При дехлорировании 1,2-дихлортетрафторэтана, например, на оксиде алюминия, модифицированном медью и оксидом хрома (673 К, время контакта 60 сек, мольное отношение хладон-114:водород=1), выход тетрафторэтилена составил 61%, а конверсия субстрата 31%.
В качестве катализаторов дехлорирования фторхлоруглеродов, содержащих 2-10 атомов углерода, предлагаются также фосфаты металлов группы железа, нанесенные на γ-Al2O3 /236,237/; количество металла составляет обычно не менее 3% от массы катализатора.
Перед реакцией катализатор обрабатывают смесью азота и фтористого водорода (мольное соотношение N2:HF=1) при 406 К. Наиболее эффективными являются никельсодержащие катализаторы; так, при дехлорировании 2,3-дихлороктафторбутана на оксиде никеля, модифицированном фосфатом никеля (926 К), был получен перфторбутен-2 с выходом 88% при степени конверсии фторхлоруглерода 92% /236/.
Описан /238/двухстадийный каталитический способ дегалаидирования фторгалоидуглеродов, содержащих 2-10 атомов углерода. На первой стадии фторгалоидирования субстрат контактируют при 480-700 К и давлений 9,8х104 – 2,0х106 Па с водородом на катализаторе, содержащем фосфаты группы железа, нанесенные на фторированную γ-Al2O3. Количество фосфата металла в катализаторе в пересчете на металл составляет 0,1-10,0 масс.%, количество фтора в носителе – 50-68 масс.%.
Образующуюся на первой стадии реакционную смесь контактируют при 480-700 К с каталитической композицией, содержащей оксиды или хлориды кобальта, никеля, цинка, хрома или железа, а также хлорид бария на носителе из активированного угля или оксида алюминия и получают фторолефины с низким содержанием побочных продуктов. Так, при дехлорировании 1,2-дихлортетрафторэтана над фторированным оксидом алюминия, модифицированным фосфатом никеля (588 К, время контакта 12,5 сек.) и последующем контактировании реакционных газов с хлоридом бария, нанесенном на активированный уголь (673 К, время контакта 180 сек) был получен тетрафторэтилен с выходом 71,2% (в одностадийном процессе его выход равнялся 58,9%). Автор /238/ полагает, что увеличение выхода целевого продукта при использовании двухстадийного способа обусловлено превращением побочных продуктов (1,1,2,2-тетрафторхлорэтана и 1,1,2,2-тетрафторэтана) на второй стадии с образованием тетрафторэтилена.
В /239/ описано получение гексафторпропилена каталитическим дегидрохлорированием 2-хлоргексафторпропана в присутствии водорода.
Процесс проводят в никелевой трубке при 650-750 К в слое смеси фторидов щелочного и щелочноземельного металла, модифицированных медью, никелем и оксидом хрома. Выход гексафторпропилена при времени контакта 2,5 сек и соотношении субстрат:водород, равном 1, составил 94%, а степень конверсии субстрата – 56%.
В /240/ приведены данные по дехлорированию 1,1,-дифтор-1,2-дихлорэтана на никелевых спиралях; при 720-870 К, мольном соотношении субстрат: водород, равном 1,25 и времени контакта 10 сек выход 1,1-дифторэтилена превышал 99%, а степень конверсии субстрата составила 40-45%.
Таким образом, для интенсификации дехлорирования фторхлоруглеродов водородом применяют нанесённые катализаторы. Активными их компонентами являются, как правило, никель, кобальт, медь или оксид хрома, а носителями – активированный уголь, силикагель, цеолиты, активный оксид алюминия, фториды алюминия, кальция и магния.
Селективность дехлорирования 1,2-дифтортетрахлорэтана, 1,1,2 –трифтортрихлорэтана и 1,2-дихлортетрафторэтана на этих катализаторах, как правило, находится на уровне 90, 75-85 и 60-65%, соответственно.
3.2.2 Дехлорирование 1,2-дихлоргексафторпропана
1,2-дихлоргексафторпропан (хладон-216) является многотоннажным отходом производства тетрафторэтилена и подвергается термическому уничтожению или выбрасываются в атмосферу. Одним из путей утилизации хладона-216 является его дехлорирование водородом с получением гексафторпропилена (ГФП) – ключевого дефицитного сырья для производства целого ряда фторорганических продуктов (фторполимеров, фторкаучуков, фторполиэфиров):
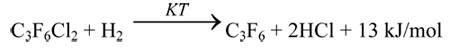
Первым этапом разработки эффективного катализатора дехлорирования 1,2-дихлоргексафторпропана был подбор каталитического носителя. В литературе отсутствуют сообщения о дехлорировании 1,2-дихлоргексафторпропана; нами были испытаны носители – активированный уголь, активный оксид алюминия, фториды алюминия, магния и кальция, которые широко используются для приготовления катализаторов однотипных процессов дехлорирования ФОС /225-240/. Кроме того, нами впервые для такого типа реакции был испытан α-Al2O3, который, как уже отмечалось, обладает повышенной стойкостью в агрессивных фторирующих средах.
Характерно, что известные носители, содержащие оксид кремния (силикагель, аморфный алюмосиликат, цеолиты), а также катализаторы на их основе разрушаются, как показали испытания, уже в первые часы эксплуатации, что объясняется, по-видимому, наличием в редакционных газах активного по отношению к оксиду кремния фтористого водорода, который является одним из побочных продуктов реакции.
Данные по каталитическим свойствам носителей приведены в табл. 28 (оп. 2-8); здесь же представлены результаты по некаталитическому взаимодействию 1,2-дихлоргексафторпропана и водорода (оп. 1).
Из приведенных данных видно, что и в отсутствии катализаторов происходит образование ГФП, однако степень конверсии субстрата невелика и даже при 843 К не превышает 10%. Видно также, что на активированном угле и γ-Al2O3 образуются значительные количества побочных продуктов, а выход целевого продукта не превышает 15%. Характерно, что химизм образования практически всех побочных продуктов можно объяснить диспропорционированием исходного 1,2-дихлоргексафторпропана и дехлорированием водородом продуктов диспропорционирования (C3F7Cl, C3F5Cl, C3F4Cl2, C3F3Cl3), а также гидрогалогенированием ГФП (C3F7H, C3F6HCl). Отметим также, что γ-Al2O3 разрушается в ходе термического контактирования с хладоном-216 и водородом.
Применение фторидов щелочноземельных металлов позволяет достичь высокого (выше 90%) выхода ГФП (оп. 5,6, табл. 28); в то же время практическое использование этих носителей весьма затруднительно, так как даже свежие образцы обладают низкой механической прочностью. Плавленый фторид кальция хотя и обладает высокой стабильной механической прочностью, характеризуется относительно низкими селективностью и активностью (оп. 7, табл. 28).
Для α-Al2O3 характерен высокий выход целевого продукта (оп. 8, табл. 28); даже длительное пребывание этого носителя в реакционной зоне в весьма жестких условиях практически не сказывается на его механической прочности. Этот результат можно объяснить по данным рентгенофазового и химического анализа α-Al2O3, а также катализаторов на его основе после эксплуатации, которые показывают образование лишь следовых количеств фазы фторида алюминия. Для катализаторов на основе γ-Al2O3, напротив, характерно глубокое его фторирование и, как следствие, потеря механической прочности.
Таким образом, испытание широкого класса носителей показало, что только α-Al2O3 обладает всеми необходимыми свойствами и может быть использован в качестве носителя для эффективного катализатора дехлорирования 1,2-дихлоргексафторпропана водородом.
Модифицирование α-Al2O3 известными активными компонентами – оксидом хрома, металлическим кобальтом, медью, а также никелем приводит во всех случаях, как следует из табл. 28 (оп. 9-12) и рис. 3а, к увеличению (по сравнению с немодифицированным α-Al2O3) степени конверсии субстрата. При этом наибольшей активностью обладает катализатор, модифицированный никелем, а наименьшей – оксидом хрома. Образцы α-Al2O3 , модифицированные кобальтом и медью имеют промежуточную активность. Выход ГФП в интервале 670-770 К на всех катализаторах примерно одинаков и находится на уровне 90 % (выход на образце, модифицированном оксидом хрома несколько ниже). При повышении температуры до 840 К выход ГФП на всех катализаторах падает.
α-Al2O3, модифицированный никелем, является весьма активным и селективным катализатором для получения ГФП дехлорированием 1,2-дихлоргексафторпропана, что предполагает возможность его практического использования; в то же время применение уже готовых промышленных гетерогенных контактов при практической реализации способа позволило бы избежать трудностей связанных с организацией производства специального катализатора. В этой связи нами был испытан промышленный никельсодержащий катализатор марки ГИАП-3-6Н, который представляет собой (после термообработки в среде водорода) никель, нанесенный на α-Al2O3, и, для сравнения, катализатор марки НКМ-І, представляющий собой никель, нанесенный на γ-Al2O3 (оп. 13-16, табл. 28).
Из приведенных данных видно, что использование катализатора ГИАП-3-6Н позволяет достичь практически количественного выхода ГФП – так, при 793 К и скорости подачи реагентов 285 час-1 выход C3F6 составляет 96,3 %, а степень конверсии 1,2-дихлоргексафторпропана – 90 % (оп. 16, табл. 28).
Аналогичный по химическому составу катализатор НКМ-І, приготовленный на основе γ-Al2O3 , характеризуется существенно более низким выходом ГФП (оп. 13, табл. 28).
Промышленный гетерогенный контакт ГИАП-3-6Н, таким образом, является эффективным катализатором дехлорирования 1,2-дихлоргексафторпропана водородом; высокий выход ГФП при этом обеспечивается использованием в качестве каталитического носителя α-Al2O3, на поверхности которого отсутствуют центры, обуславливающие протекание побочных реакций.
По результатам работы выданы исходные данные на проектирование опытно-промышленной установки по утилизации хладона-216 с получением ГФП.
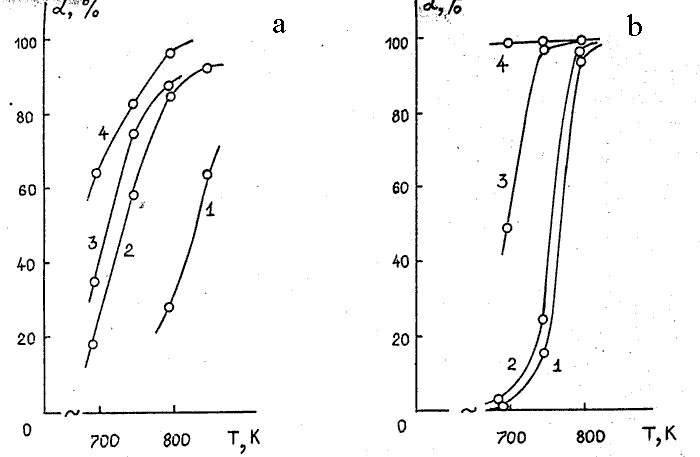
Рисунок 3. Температурные зависимости степеней конверсии (α) 1,2-дихлоргексафторпропана (а) и 1,2-дифтортетрахлорэтана (б) при контактировании с водородом на катализаторах: 1 – α-Al2O3; 2 – Co /α-Al2O3; 3 – Сu /α-Al2O3; 4 – Ni /α-Al2O3 (объёмная скорость подачи эквимолярной смеси реагентов 285 час -1).
3.2.3 Дехлорирование 1,2-дифтортетрахлорэтана.
1,2-дифтордихлорэтилен явялется исходным реагентом для синтеза перфторметилперфторвинилового эфира - сырья для производства новых типов фторкаучуков и фторполимеров. Одним из путей получения
1,2-дифтордихлорэтилена является каталитическое дехлорирование водородом 1,2-дифтортетрахлорэтана:
![]()
Данные по каталитическому дехлорированию 1,2-дифтортетрахлорэтана водородом приведены в табл. 29; здесь же представлены результаты по их некаталитическому взаимодействию.
Видно, что при температуре ниже 740 К реакция в отсутствии катализатора (пустотелый кварцевый реактор) практически не протекает, конверсия субстрата в этих условиях не превышает 6 %. При температурах выше 740 К наблюдается резкое увеличение степени конверсии даже в отсутствие катализатора (оп. 2, табл. 29); выход целевого продукта – 1,2-дифтордихлорэтилена при этом невелик и находится на уровне 60 %.
Анализ данных по каталитическим свойствам известных носителей (γ-Al2O3; AlF3, активированный уголь, MgF2) показывает, что наименьшей селективностью обладает γ-Al2O3 – выход 1,2-дифтордихлорэтилена в этом случае не превышает 2 %, а основными продуктами реакции являются хладоны-113, -114 и тетрахлорэтилен.
Данные по каталитическим свойствам фторида алюминия (получен фторированием γ-Al2O3 элементарным фтором), активированного угля и фторида магния показывают, что выход целевого продукта на этих носителях относительно высок и составляет 80-90 % при степенях конверсии 1,2-дифтортетрахлорэтана – 70-99 % (оп. 5-10, табл. 29). В то же время практическое использование этих носителей весьма затруднительно. Фториды алюминия и магния имеют низкую механическую прочность и разрушаются в ходе эксплуатации. Активированный уголь постепенно теряет активность; в то же время регенерация высокотемпературной обработкой воздухом, эффективная для минеральных носителей /224/, в случае активированного угля совершенно неприемлема.
Максимальный выход целевого продукта при испытании носителей был достигнут на α-Al2O3 – 94,3 % при полной конверсии субстрата (оп. 12, табл. 29). Испытания показали, что α-Al2O3 обладает повышенной механической прочностью, которая не уменьшается даже при длительной его эксплуатации. Характерной особенностью α-Al2O3 является отсутствие образования на его поверхности продуктов уплотнения, которые могут снижать каталитическую активность. Все это позволяет считать α-Al2O3 наиболее подходящим носителем для эффективного катализатора дехлорирования 1,2-дифтортетрахлорэтана, а также использовать как катализатор и в немодифицированном виде.
Характерно, что химизм образования практически всех побочных продуктов взаимодействия 1,2-дифтортетрахлорэтана и водорода, как и для случая 1,2-дихлоргексафторпропана, можно объяснить диспропорционированием исходного субстрата и дехлорированием водородом продуктов диспропорционирования (С2F5Cl, С2F4Cl2 , С2F3Cl3 , С2Cl4 , С2FCl3, С2FCl5), а также гидрогалогенированием образующихся при этом галоидолефинов (С2F4ClН, С2F2Cl3Н). Кроме того, при взаимодействии 1,2-дифтортерахлорэтана с водородом образуются продукты замещения хлора в молекуле 1,2-дифтордихлорэтилена на водород – 1,2-дифторхлорэтилен и 1,2-дифторэтилен (выход С2F2Н2, в оп. 2, например, составил 4,2 %).
Интересно, что α-Al2O3 весьма инертен, как показали испытания, в реакциях диспропорционирования; так, степень конверсии 1,2-дифтортетрахлорэтана на этом носителе в среде азота при повышенных температурах – 843 и 893 К составила всего 1,2 и 3,2 %, соответственно. В продуктах реакции при этом были обнаружены 1,2-дифтордихлорэтилен и хлор – это указывает на протекание дехлорирования и в инертной среде. Роль водорода при дехлорировании фторхлоруглеродов на носителях (особенно при высоких температурах), таким образом, может заключаться в связывании образующегося хлора и в соответствующем сдвиге равновесия диссоциации субстрата на фторолефин и хлор.
Модифицирование α-Al2O3 оксидом хлора, кобальтом, медью и никелем приводит, как видно из результатов, представленных в табл. 29 (оп. 13-20) и на рис. 3б, к существенному росту активности, которая увеличивается в ряду Co<Cr2O3<Cu<Ni. Для контактов, модифицированных никелем, полная конверсия субстрата наблюдается уже при 690 К, медью при 740 К, а кобальтом и оксидом хрома – только при 790 К.
Наименьшей селективностью обладают контакты, модифицированные оксидом хрома; основными побочными продуктами здесь являются 1,1,2-трифтортрихлорэтан и пентафторхлорэтан, что указывает на существенный вклад диспропорционирования субстрата. Наибольшей селективностью обладает α-Al2O3, модифицированный никелем; выход 1,2-дифтордихлорэтилена при полной конверсии 1,2-дифтортетрахлорэтана здесь достигает 97,1 % (оп. 19, табл. 29). Столь высокие активность и селективность действия катализатора Ni /α-Al2O3 создают предпосылки для промышленной реализации дехлорирования хладона-112 водородом.
Испытание промышленного катализатора ГИАП-3-6Н (Ni/α-Al2O3) выявили его высокую активность и селективность (оп. 23-25, табл. 29); выход 1,2-дифтордихлорэтилена при практически полной конверсии субстрата достигал 98,5%. Испытанный для сравнения аналогичный по химическому составу промышленный никельсодержащий катализатор НКМ-1, приготовленный на основе γ-Al2O3, обладает существенно меньшей селективностью действия (оп. 21, табл. 29) и разрушается в ходе эксплуатации. Кроме побочных продуктов, приведенных табл. 29, для этого катализатора характерно образование значительных количеств 1,2-дифторэтилена, выход которого достигал 24,8% (693 К). Интересно, что предварительная обработка катализатора НКМ-1 фтором (743 К) приводит к существенному увеличению селективности его действия (оп. 22, табл. 29).
Отличительной особенностью катализатора ГИАП-3-6Н является высокая механическая прочность, которая не уменьшается при эксплуатации и регенерации. Длительная эксплуатация этого катализатора сопровождается снижением его активности, которое обусловлено образованием на поверхности продуктов уплотнения; регенерация воздухом (770-820 К) приводит к полному восстановлению каталитической активности.
С целью увеличения стабильности дехлорирования 1,2-дифтортетрахлорэтана водородом нами был испытан двухслойный катализатор. Первым слоем являлся немодифицированный α-Al2O3 (80 масс.%), вторым – катализатор ГИАП-3-6Н (20 масс.%). Такой двухслойный катализатор показал высокую активность и селективность. Так, при 740-760 К наблюдалась полная конверсия субстрата, а селективность процесса достигала 97%. Двухслойный катализатор обладал, кроме того, повышенной стабильностью. Этот эффект обусловлен, по-видимому, тем, что в первых слоях, то есть там, где коксообразование протекает наиболее интенсивно, использовался α-Al2O3, на котором продукты уплотнения не образуются.
По результатам работы выданы исходные данные на проектирование технологии получения 1,2-дифтордихлорэтилена дехлорированием 1,2-дифтортетрахлорэтана водородом на двухслойном катализаторе α-Al2O3 – ГИАП-3-6Н.
3.2.4 Дехлорирование 1,1,2-трифтортрихлорэтана.
Производство трифторхлорэтилена – сырья для синтеза фторопластов и фторкаучуков, основано на дехлорировании 1,1,2-трифтортрихлорэтана дорогим и дефицитным металлическим цинком и характеризуется, как уже отмечалось, низкой производительностью, высокой материалоемкостью, а также загрязнением окружающей среды токсичным хлоридом цинка. Всех этих недостатков можно избежать, осуществляя дехлорирование 1,1,2-трифтортрихлорэтана водородом:
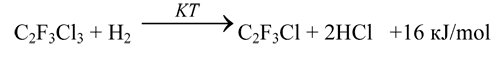
Данные по каталитическим свойствам известных носителей для катализаторов дехлорирования фторхлоруглеродов водородом – γ-Al2O3, активированного угля, фторидов алюминия, магния, а также плавленого фторида кальция приведены в табл. 30; здесь же представлены результаты, полученные с использованием α-Al2O3 и, для сравнения – в полом реакторе, без катализатора.
Видно, что даже в отсутствии катализатора наблюдается значительная конверсия 1,1,2-трифтортрихлорэтана; селективность дехлорирования при этом снижается с увеличение температуры (оп. 1,2, табл. 30). Так, при 793 и 843К степень конверсии субстрата составила 11 и 88%, а выход трифторхлорэтилена – 78,3 и 53,7%, соответственно. Основным побочным продуктом при некаталитическом газофазном взаимодействии 1,1,2-трифтортрихлорэтана и водорода является трифторэтилен, выход которого при 843 К, например, составил 24,8%.
Анализ данных, полученных с использованием γ-Al2O3 и AlF3 (оп. 3-6, табл. 30) показывает, что основными здесь являются продукты диспропорционирования исходного хладона-113 – хладоны -115, -114, -112, а также продукт дехлорирования гексахлорэтана – тетрахлорэтилен. Выход целевого продукта при этом не превышает 0,1%. Кроме того, в продуктах реакции были обнаружены хладоны -122, -123, -124 и 1,2-дифтордихлорэтилен. Интересно отметить, что эти же продукты образуются при дехлорировании 1,2-дифтортетрахлорэтана.
При использовании в качестве катализатора активированного угля выход целевого продукта значительно выше и достигает 89,8% (оп. 7,8, табл. 30); существенным недостатком активированного угля является то, что его активность резко уменьшается во времени, а регенерация высокотемпературной обработкой воздухом здесь неприемлема.
В дальнейшем нами были испытаны фториды магния и кальция. Фторид магния был приготовлен прессованием высокодисперсной соли (поверхность – 12,5 м2/г); этот образец характеризовался низкой механической прочностью. Напротив, фторид кальция был весьма прочен; его получали методом плавления (поверхность 0,2м2/г).
Из табл. 30 (оп. 9-11) видно, что степень конверсии субстрата на относительно более дисперсном фториде магния существенно выше. Основные побочные продукты здесь такие же, как и при некаталитическом взаимодействии 1,1,2-трифтортрихлорэтана с водородом – трифторэтилен и изомеры трифтордихлорэтана (хладона-123); выход целевого продукта относительно высок и находится на уровне 85-90%.
Анализ данных, полученных с использованием α-Al2O3 показывает, что применение этого носителя позволяет достичь высокого выхода трифторхлорэтилена в широком интервале температур; так при 740-790 К выход целевого продукта находится на уровне 86-91% (оп. 12,13, табл. 30). Интересно, что активность этого носителя даже в немодифицированном виде весьма велика (выше, чем у MgF2 и CaF2); ранее нами уже отмечалось, что α-Al2O3 является эффективным катализатором дехлорирования 1,2-дифтортетрахлорэтана.
Испытание каталитических носителей выявило, таким образом, что удовлетворительной стабильной механической прочностью обладают только плавленый фторид кальция и α-Al2O3.
Модифицирование фторида кальция медью и, особенно, никелем приводит к существенному увеличению каталитической активности (оп. 14-17, табл. 30), выход целевого продукта при этом составляет 87-90%.
α-Al2O3, модифицированный оксидом хрома, характеризуется значительным образованием хладона-114 и дифтодихлорэтилена; суммарный выход этих побочных продуктов достигает 23% (оп. 18,19, табл. 30).
Применение α-Al2O3, модифицированного палладием, приводит к существенному увеличению выхода трифторэтилена (оп. 20, табл. 30).
α-Al2O3, модифицированный кобальтом, при температурах ниже 740 К характеризуется высокой селективностью (93-95%); степень конверсии субстрата при этом невелика (ниже 25%). Повышение температуры до 793 К сопровождается (оп. 22, табл. 30) заметным снижением селективности (до 85,7%).
Модифицирование α-Al2O3 никелем приводит к резкому увеличению каталитической активности при умеренных температурах (690-740 К).Так, степень конверсии 1,1,2-трифтортрихлорэтана на α-Al2O3 после его модифицирования никелем возрастает при 693 К от 2 до 41%, а при 743 К – от 23 до 70%. (оп. 23,24, табл. 30).
Селективность дехлорирования на катализаторе Ni/α-Al2O3, в том числе, и на промышленном образце ГИАП-3-6Н (оп. 25,26, табл. 30) составляет 84-90% при степенях конверсии 1,1,2-трифтортрихлорэтана – 40-90%. Для сравнения, выход целевого продукта на катализаторе Ni/γ-Al2O3 (НКМ-1) в аналогичных условиях составил всего 19,5% (оп. 27, табл. 30).
Резкое увеличение селективности дехлорирования 1,1,2-трифтортрихлорэтана (94%) было выявлено нами при изучении каталитических свойств α-Al2O3, модифицированного медью (оп. 28-30, табл. 30). Начальная активность этого катализатора также велика – степень конверсии субстрата на свежих образцах при температуре выше 790 К превышает 99%.
Особенностью Сu/α-Al2O3 является заметное снижение его активности в ходе опыта (сравни оп. 29,30, табл. 30). Регенерация термообработкой в токе воздуха, которая приводит к практически полному восстановлению активности никельсодержащих катализаторов, для Сu/α-Al2O3 оказалась малоэффективной. Анализ состава этого катализатора до и после его эксплуатации в течение 80 часов (793 К) показал существенное снижение содержания меди (с 9,8 до масс. 2,8%). Таким образом, низкая стабильность Сu/α-Al2O3 может быть обусловлена его обеднением активным компонентом, например, вследствие уноса в виде летучего хлорида меди. Активность катализатора может также снижаться при уменьшении дисперсности нанесенной меди.
Известным приемом повышения стабильности медьсодержащих катализаторов дехлорирования фторхлоруглеродов водородом является введение промотирующих добавок – оксидов хрома и бария /225, 228, 233-235, 237/.
Анализ данных, полученных с использованием приготовленного нами катализатора Cu,BaO,Cr2O3/α-Al2O3, свидетельствует о существенном уменьшении селективности процесса по сравнению с непромотированным Cu/α-Al2O3 (оп. 31, табл. 30), в основном за счет образования дихлортетрафторметана и дифтордихлорэтилена, характерных и для катализатора Cr2O3/α-Al2O3.
В дальнейшем нами был приготовлен и испытан катализатор Cu,BaO/α-Al2O3, где в качестве промотора использовался только оксид бария (2 масс.%). Видно (оп. 32-35, табл. 30), что даже при 793 К этот образец характеризуется исключительно высокой селективностью действия, которая достигает 96,1%. Стабильная активность устанавливается примерно через 2-4 часа после начала эксплуатации и отвечает при 793 К степени конверсии субстрата, равной 74-76%; эксплуатация катализатора в течение 53 часов не привела к уменьшению его активности.
Активность медьсодержащих катализаторов на основе плавленого фторида кальция, который также как и α-Al2O3 устойчив в условиях осуществления процесса, существенного ниже, по-видимому, из-за малой удельной поверхности носителя (0,2 м2/г), что приводит, как следствие, к снижению выхода целевого продукта из-за большего вклада малоселективного некаталитического дехлорирования субстрата водородом (например, между зерен катализатора).
Таким образом, использование устойчивого во фторирующих средах и обладающего относительно развитой поверхностью α-Al2O3 (7,5-8,0 м2/г) позволило подобрать эффективный катализатор дехлорирования 1,1,2-трифтортрихлорэтана водородом – Cu,BaO/α-Al2O3.
По результатам работы выданы исходные данные на проектирование полупромышленной установки по получению трифторэтилена дехлорированием хладона-113 водородом.
3.2.5 Дехлорирование 1,2-дихлортетрафторэтана.
Одним из перспективных путей получения тетрафторэтилена является каталитическое дехлорирование 1,2-дихлортетрафторэтана (хладона-114) водородом
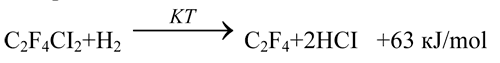
В табл. 31 приведены данные по дехлорированию 1,2-дихлортетрафторэтана водородом на подобранных нами эффективных катализаторах однотипных процессов дехлорирования хладонов-216, -112 и -113; общей чертой этих катализаторов – Ni/α-Al2O3 и Cu,BaO/α-Al2O3 является то, что они приготовлены на основе α-Al2O3. Для сравнения здесь же приведены данные, полученные с использованием никеля, диспергированного на поверхности известного носителя для катализатора дехлорирования фторхлоуглеродов – γ-Al2O3/238/.
Таблица 31. Каталитическое дехлорирование 1,2-дихлортетрафторэтана водородом (Скорость подачи эквимолярной смеси реагентов – 285 час-1)
|
# оп. |
Катализатор |
Температура К |
Степень конверсии фторхлор-углерода, % |
Выход продуктов реакции, % |
||||
|
C2F6 |
C3F8 |
C4F10 |
C5F12 |
Прочие |
||||
|
1 |
α-Al2O3 |
823 |
26 |
52,5 |
0,8 |
3,2 |
16,7 |
26,8 |
|
2 |
То же |
873 |
67 |
48,9 |
1,5 |
6,8 |
17,1 |
25,7 |
|
3 |
Ni/α-Al2O3 |
773 |
33 |
64,7 |
1,1 |
3,9 |
12,5 |
17,8 |
|
4 |
То же |
823 |
48 |
61,8 |
2,2 |
5,2 |
14,2 |
16,6 |
|
5 |
Cu,BaO/ α-Al2O3 |
823 |
43 |
75,0 |
1,7 |
4,4 |
8,2 |
10,7 |
|
6 |
То же |
873 |
85 |
65,7 |
1,7 |
4,5 |
8,5 |
19,6 |
|
7 |
NiF2/ γ-Al2O3 |
773 |
62 |
21,5 |
2,6 |
8,4 |
31,7 |
35,8 |
|
8 |
То же |
823 |
78 |
14,7 |
3,5 |
12,4 |
30,5 |
38,9 |
Из приведенных данные видно, что дехлорирование 1,2-дихлотетрафторэтана характеризуется более низким выходом целевого продукта и протекает в существенно более жестких условиях (по сравнению с хладонами-112,-113 и -216). Максимальный выход тетрафторэтилена (75%) был достигнут на катализаторе Cu,BaO/α-Al2O3 при 823 К; степень конверсии субстрата составила при этом 43% (оп. 5, табл. 31). Кроме тетрафторэтилена при взаимодействии хладона-114 с водородом образуются значительные количества трифторхлорэтилена и дифтордихлорэтилена, по-видимому, при дехлорировании хладонов-113 и -112 – продуктов диспропорционирования исходного субстрата. В реакционных газах были обнаружены также гексафторэтан, пентафторхлорэтан и тетрахлорэтилен.
Характерно, что использование катализатора на основе γ-Al2O3 (оп. 7,8, табл. 31) приводит к резкому увеличению вклада побочных процессов и соответствующему снижению выхода тетрафторэтилена.
3.2.6 Дехлорирование перфторметокси-1,2-дихлортрифторэтана.
Каталитическое дехлорирование водородом перфторметокси-1,2-дихлортрифторэтана является одним из возможных вариантов получения перфторметилперфторвинилового эфира – мономера для синтеза новых типов фторкаучуков и фторполимеров:

Данные по каталитическому дехлорированию перфторметокси-1,2-дихлортрифторэтана приведены в табл. 32.
Из приведенных данных видно, что селективность дехлорирования перфторметокси-1,2-дихлортрифторэтана водородом невелика; максимальный выход целевого продукта (42,6%) был достигнут на катализаторе Ni/α-Al2O3 при 573К, степень конверсии субстрата составила при этом 15% (оп. 5, табл. 32).
Таблица 32. Каталитическое дехлорирование перфторметокси-1,2-дихлортрифторэтана водородом (скорость подачи эквимолярной смеси реагентов – 285 час-1).
|
# оп. |
Катализатор |
Температура К |
Степень конверсии, % |
Выход продуктов реакции, % |
|||||
|
CF3OC2F3 |
C2F4Cl2 |
CF3Cl |
CF3H |
C2F3Cl |
Прочие |
||||
|
1 |
α-Al2O3 |
623 |
3 |
24,3 |
2,5 |
10,4 |
16,7 |
10,5 |
35,6 |
|
2 |
То же |
673 |
13 |
17,0 |
3,8 |
18,6 |
22,4 |
16,9 |
21,3 |
|
3 |
Cu,BaO/α-Al2O3 |
573 |
10 |
36,4 |
4,0 |
6,2 |
11,0 |
11,3 |
31,1 |
|
4 |
То же |
623 |
18 |
18,9 |
4,8 |
7,1 |
12,4 |
12,5 |
44,3 |
|
5 |
Ni/α-Al2O3 |
573 |
15 |
42,6 |
3,5 |
9,0 |
11,7 |
8,7 |
24,5 |
|
6 |
То же |
573 |
57 |
22,9 |
4,3 |
10,3 |
26,0 |
14,8 |
21,7 |
|
7 |
Ni/γ-Al2O3 |
573 |
34 |
11,6 |
36,1 |
20,5 |
13,3 |
5,8 |
12,7 |
|
8 |
То же |
623 |
87 |
7,4 |
38,5 |
22,4 |
14,2 |
7,5 |
10,0 |
Основным побочным процессом яляется деструкция исходного фторхлоруглерода с образованием 1,2-дихлортетрафторэтана, трифторхлорэтилена, трифторметана и карбонилдифторида. Ni/ γ-Al2O3 характеризуется (оп. 7,8, табл. 32) повышенным вкладом деструкции сырья (по сравнению с аналогичными катализаторами на основе α-Al2O3); деструкция интенсивно протекает и в отсутствии водорода.
3.2.7 Общие закономерности, кинетика и механизм каталитического дехлорирования фторхлоруглеродов водородом.
Проведенное исследование дехлорирования фторхлоруглеродав водородом показало определяющее влияние природы гетерогенного катализатора (наряду с природой субстрата) на селективность и скорость процесса. Селективность дехлорирования определяется, в основном, характером каталитического носителя, а скорость – его удельной поверхностью и природой активного металлического компонента каталитической композиции.
Механизм дехлорирования фторхлоруглеродов водородом на нанесенных металлических катализаторах может включать две стадии: взаимодействие фторхлорулерода (RCl2) и металла (Ме) с образованием соответствующего галоидолефина (R:), а также хлорида металла (MeCl2) и, на второй стадии, восстановление хлорида металла водородом с регенерацией металлических каталитических центров и образованием хлористого водорода:
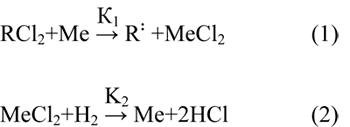
(К1 и K2 – константы скоростей соответствующих реакций).
Для этого маршрута при стационарной концентрации свободных металлических центров (θМе) дехлорирование фторхлоруглеродов водородом описывается следующим кинетическим уравнением:
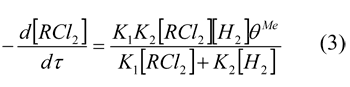
или, при малых степенях конверсии фторхлоруглерода (αRCl2) и водорода, уравнением:
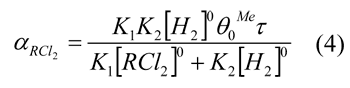
где [RCl2]° и [H2]° - начальные концентрации фторхлоруглерода и, соответственно, водорода θ0Ме – начальная концентрация металлических активных центров катализатора, τ – время контакта реакционных газов с катализатором.
Возможны и другие маршруты реакции; выше отмечалось, что дехлорирование может включать, например, каталитическую диссоциацию фторхлоруглерода на галоидолефин, а также элементарный хлор и последующее связывание хлора водородом.
Для уточнения механизма взаимодействия фторхлоруглеродов и водорода на нанесенных металлических катализаторах нами были детально, на примере 1,2-дихлоргексафторпропана и катализатора Ni/α-Al2O3, изучены в сопоставимых условиях кинетические закономерности
- взаимодействия субстрата с металлическим никелем, нанесенным на α-Al2O3;
- восстановления водородом хлорида никеля, нанесенного на α-Al2O3;
- взаимодействия субстрата и водорода в слое никеля, нанесенного на α-Al2O3.
Такое комплексное изучение позволяет определять константы К1 и К2, характеризующие кинетику «полуреакций» (1) и (2), независимо; сопоставление их значений с определенными непосредственно при исследовании кинетики дехлорирования субстрата водородом, делает возможным количественную оценку вклада данного маршрута. Исследование кинетики взаимодействия 1,2-дихлоргексафторпропана с никелем, нанесенным на α-Al2O3 и результаты изучения кинетики восстановления водородом хлорида никеля, нанесенного на α-Al2O3 позволили определить начальную концентрацию активных никеля и хлорида никеля на поверхности α-Al2O3 (θ0Ni , θ0NiCl2), и по их температурной зависимости определить соответствующие значения предэкспоненциальных множителей (Ко) и эффективных энергий активаций (Еа); результаты представлены в табл. 33 (п.п. 1-4) и на рис. 4а.
Таблица 33. Значения начальных концентраций активных никеля и его хлорида на поверхности
α-Al2O3, а также констант скоростей К1 и К2, определенных
прямыми измерениями (пп. 1-4) и из кинетических данных по дехлорировнаию 1,2-дихлоргексафторпропана
водородом (пп. 5-6).
|
# оп. |
Определяемая величина |
Значения определяемой величины при температуре, К |
lgKo |
Еа, кДж/ моль |
||||
|
473 |
493 |
508 |
523 |
543 |
|
|
||
|
1 |
К1, л•моль-1•сек-1, х102 |
5,9 |
7,4 |
8,9 |
10,3 |
12,9 |
1,4±0,1 |
23,8±0,5 |
|
2 |
θ0Ni, моль•л-1 |
1,13 |
1,12 |
1,15 |
1,17 |
1,17 |
- |
- |
|
3 |
К2, л•моль-1•сек-1, х102 |
1,5 |
3,3 |
5,9 |
11,6 |
22,8 |
7,4±0,2 |
84,1±2,1 |
|
4 |
θ0NiCl2, моль• л-1 |
1,24 |
1,17 |
1,15 |
1,13 |
1,06 |
- |
- |
|
5 |
К1,л•моль-1•сек-1, х102 |
5,7 |
9,1 |
11,0 |
14,5 |
19,1 |
2,8±0,2 |
36,4±1,7 |
|
6 |
К2, л•моль-1•сек-1, х102 |
1,3 |
2,9 |
5,9 |
11,3 |
26,8 |
8,4±0,3 |
93,4±2,6 |
Из приведенных данных видно, что взаимодействие 1,2-дихлоргексафторпропана и металлического никеля на поверхности α-AI2O3 характеризуется относительно небольшой энергией активации (23,8 кДж/моль), которая существенно ниже энергии активации восстановления водородом хлорида никеля на поверхности α-AI2O3 (84,1 кДж/моль); в то же время предэкспоненциальный множитель константы скорости восстановления хлорида никеля водородом существенно выше, чем у константы дехлорирования субстрата никелем. Если учесть, что начальные концентрации активных никеля и хлорида никеля в образцах Ni/α-Al2O3 и NiCl2/α-AI2O3, примерно одинаковы (~ 1,15 моль/л образца), то, по совокупности, при температурах ниже 520 К скорость дехлорирования субстрата никелем выше скорости восстановления хлорида никеля водородом (рис. 4а) и, напротив, при повышенных температурах, в соответствии с данными по изучению кинетики модельных реакций (1) и (2), дехлорированние 1,2-дихлоргексафторпропана никелем является лимитирующей стадией.
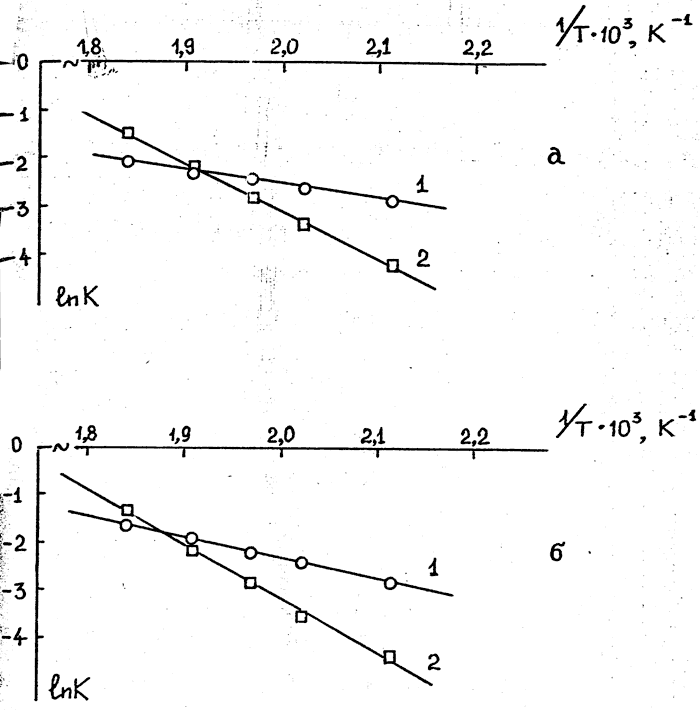
Рисунок 4. Температурные зависимости (в аррениусовых координатах) констант скоростей дехлорирования 1,2-дихлоргексафторпропана никелем, нанесенным на α-AI2O3 (1), и восстановление водородом хлорида никеля, нанесенного на α-AI2O3 (2), определенных прямыми измерениями (а) и из кинетических данных по каталитическому дехлорированию 1,2-дихлоргексафторпропана водородом на Ni/α-AI2O3 (б).
Выражение (4), характеризующее кинетику дехлорирования фторхлоруглеродов водородом (при малых степенях конверсии) по предлагаемому механизму можно представить в виде:
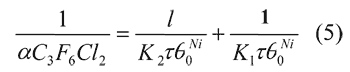
где l – мольное отношение C3F6Cl2 и H2 в исходной смеси.
Таким образом, обрабатывая кинетические данные по дехлорированию фторхлоруглеродов водородом в координатах 1/α ÷ l и используя независимо определенное значение θ0Ni (1,15 моль/л катализатора), можно непосредственно определять численные значения К1 и К2.
Результаты обработки кинетические данных по дехлорированию 1,2-дихлоргексафторпропана водородом на катализаторе Ni/α-Al2O3 представлены на рис. 4, а также в табл. 33 (пп. 5, 6).
Из приведенных данных видно, что кинетика дехлорирования 1,2-дихлоргексафторпропана водородом на Ni/α-Al2O3 удовлетворительно описывается кинетическим уравнением (5). Характерно, что температурные зависимости констант К1 и К2 и в этом случае пересекаются (рис. 4б); при температурах выше 520 К лимитирующей является стадия дехлорирования субстрата никелем и, наоборот, при пониженных температурах, константа скорости дехлорирования субстрата никелем существенно выше константы скорости восстановления водородом образующегося хлорида никеля. Этот результат полностью согласуется с закономерностями, выявленными при изучении кинетики модельных реакций (1, 2) и свидетельствует в пользу описываемого нами механизма дехлорирования 1,2-дихлоргексафторпропана водородом.
На рис. 5 приведена экспериментально определенная температурная зависимость степени конверсии 1,2-дихлоргексафторпропана при термическом контактировании с водородом на катализаторе Ni/α-Al2O3; здесь же приведена аналогичная зависимость, полученная расчетным путем на основании значений независимо определенных констант К1 и К2 модельных реакций (1) и (2). Из приведенных данных видно, что эти константы удовлетворительно описывают (в интервале температур 470-550 К) кинетику дехлорирования 1,2-дихлоргексафторпропана – расхождение экспериментальных данных с расcчётными не превышает 25%. Этот результат однозначно указывает на то, что маршрут дехлорирования
1,2-дихлоргексафторпропана водородом на Ni/α-AI2O3, описываемый уравнениями (1) и (2), в интервале 470-550 К, является основным.
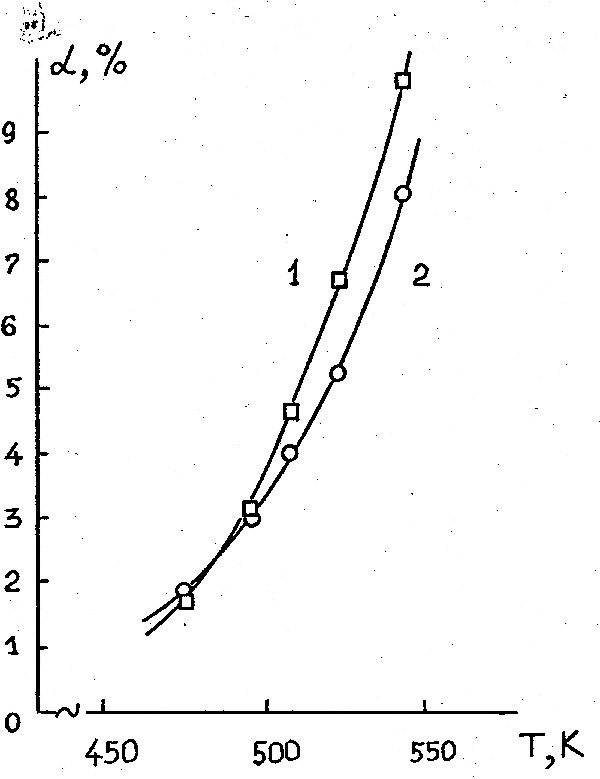
Рисунок 5. Температурные зависимости степени конверсии (α) 1,2- дихлоргексафторпропана при взаимодействии с водородом на Ni/α-AI2O3, полученные экспериментально (1) и расчетом (2) на основании значений независимо определенных констант дехлорирования 1,2-дихлоргексафторпропана никелем, нанесенным на α-AI2O3, и восстановления водородом хлорида никеля, нанесенного на α-AI2O3.
Рассматриваемая модель позволяет количественно (на основе найденных значений констант К1 и К2) оценить температурную зависимость стационарных долей металлических центров и хлорида металла (θNi / θ0Ni и θNiCl2/θ0Ni) на поверхности Ni/α-AI2O3:
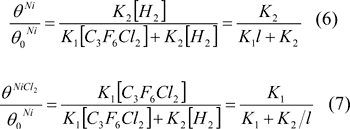
где
![]()
На рис.6 приведены температурные зависимости стационарных долей свободных активных металлических центров и центров, образовавших хлорид никеля на поверхности Ni/α-AI2O3 (для мольного соотношения субстрат:водород, равного 0,32), рассчитанные на основе значений К1 и К2, определенных из данных по кинетике дехлорирования 1,2- дихлоргексафторпропана. Характер этих зависимостей объясняется изменением соотношения значений К1 и К2 с увеличением температуры реакции. Так, в области низких температур (400-450 К) константа скорости дехлорирования субстрата никелем (К1) существенно выше константы скорости восстановления водородом, образующегося при этом хлорида никеля (К2) – как следствие, в стационарном состоянии большая доля активной поверхности катализатора представляет собой хлорид никеля. Напротив, при увеличении температуры (до 550-600 К), когда резко возрастает скорость восстановления хлорида никеля водородом, стационарная поверхность активной фазы представляет собой в основном металлический никель.
Отметим, что этот вывод, вытекающий из характера температурных зависимостей скоростей отдельных стадий дехлорирования 1,2-дихлоргексафторпропана водородом, был подтверждён нами экспериментально: рентгенофазовый анализ образцов Ni/α-AI2O3 после контакта со смесью субстрата и водорода при 400 и 600 К, соответственно, обнаружил (кроме фаз α-AI2O3и AlF3) в первом случае только фазу хлорида никеля, а во втором – только металлический никель.
Изучение реакционной способности 1,2-дифтортетрахлорэтана, 1,2- дихлоргексафторпропана, 1,1,2-трифтортрихлорэтана и 1,2-дихлортетрафторэтана в сопоставимых условиях показало, что она неодинакова и коррелирует с тепловым эффектом реакции. Так, тепловой эффект дехлорирования водородом в этом ряду субстратов составляет – 13, +13, +16 и +63 кДж/моль, соответственно; в этом же ряду (рис. 7) наблюдается существенное уменьшение реакционной способности фторхлоруглеродов.
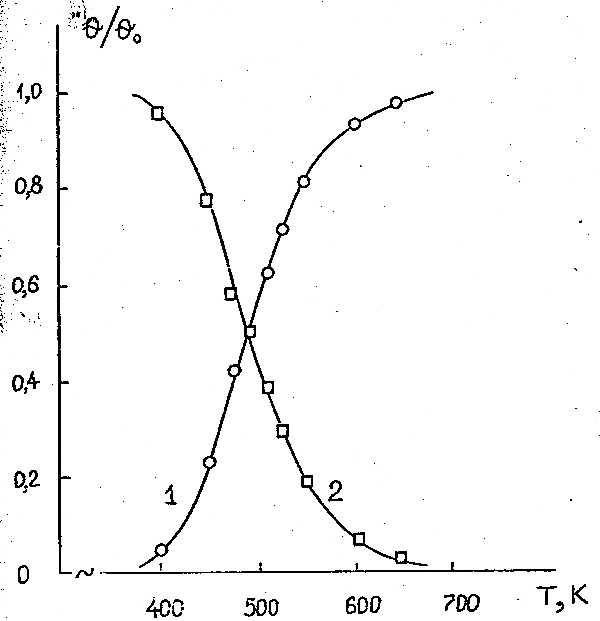
Рисунок 6. Температурные зависимости стационарных долей (Ө / Ө0) свободных активных металлических центров (1) и центров, образовавших хлорид никеля (2) на поверхности Ni /α-AI2O3 при дехлорировании 1,2-дихлоргексафторпропана водородом.
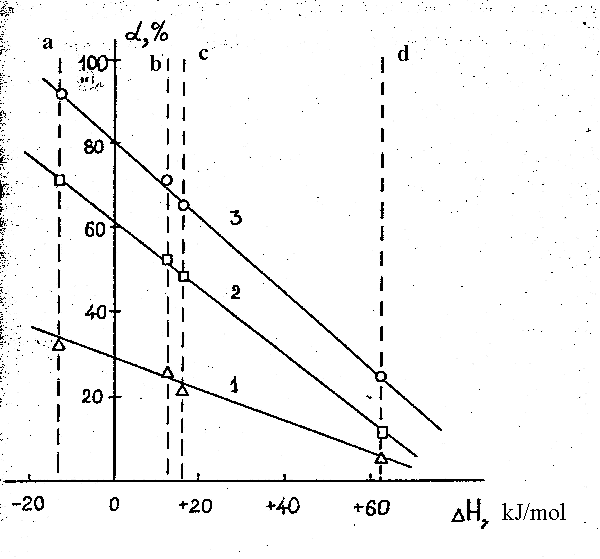
Рисунок 7. Корреляция начальных степеней конверсии фторхлоруглеродов (α) при взаимодействии
с водородом на Ni /α-AI2O3 при 1 – 573; 2 – 623 и 3 – 673 К с тепловыми
эффектами этих реакций (ΔН) для
а – 1,2-дифтортетрахлорэтана,
b – 1,2-дихлоргексафторпропана,
c– 1,1,2-трифтортрихлорэтана и
d – 1,2- дихлортетрафтоэтана.
Этот результат может быть обусловлен повышенным сродством двойной связи к хлору у галоидолефинов, содержащих большее число атомов фтора у двойной связи, что объясняется положительным мезомерным эффектом фтора; так, в соответствии с /319/, тепловой эффект хлорирования С2Сl4 , С2FСl3 , С2F2Сl2 , С3F6 , С2F3Сl и С2F4 составляет -127, -158, -171, -197, -200 и -247 кДж/моль, соответственно.
Полученные данные согласуются с рассматриваемой двухстадийной моделью, объясняющей химизм образования целевых продуктов дехлорирования; действительно, эта модель включает стадию взаимодействия фторхлоруглерода с нанесенным металлом, скорость которой в значительной степени должна определяться реакционной способностью органического субстрата.
Химизм образования побочных продуктов при каталитическом дехлорировании фторхлоруглеродов водородом.
Анализ состава реакционных смесей, образующихся при термическом контактировании фторхлоруглеродов и водорода на катализаторах (табл. 28-32), позволил предложить общую для всех субстратов схему, объясняющую химизм образования практически всех основных побочных продуктов; ниже эта схема конкретизирована для изученных реакций.
Дехлорирование 1,2-дихлоргексафторпропана:
Дехлорирование 1,2-дифтортетрахлорэтана. 1,1,2-трифтортрихлорэтана и 1,2-дихлортетрафторэтана:
Эта схема, по сути, основана на двух альтернативных механизмах образования побочных продуктов. Первый включает диспропорционирование исходного фторхлоруглерода и дехлорирование водородом продуктов диспропорционирования. Этот механизм основан на экспериментально установленном факте диспропорционирования фторхлоруглеродов в инертной среде (азот) на изучаемых катализаторах; образующиеся при этом продукты с увеличенным содержанием хлора (правая часть схемы) реагируют c водородом, как уже отмечалось, даже более интенсивно, чем исходные фторхлоруглероды.
Второй, менее очевидный механизм образования побочных продуктов включает каталитическое взаимодействие целевого галоидолефина с хлористым водородом и фтористым водородом (присоединение по двойной связи), образующимися при взаимодействии фторхлоруглерода и водорода, и последующую цепочку реакций гидро-дегидрогалогенирования (см. схему).
Для обоснования этого механизма и оценки вкладов гидро-дегидрогалогенирования и диспропрционирования нами были изучены (в качестве модельных) превращения 1,1,1-дифторхлорэтана на γ-Al2O3 в импульсном и проточном режимах. Особенность протекания процесса в импульсном режиме заключается в хроматографическом разделении продуктов первичных реакций (1,1-дифторэтилена, 1,1-фторхлорэтилена, фтористого и хлористого водорода) на слое катализатора. Такое разделение препятствует протеканию вторичных реакций гидрогалогенирования с образованием, в частности, 1,1,1-трифторэтана и не влияет на диспропорционирование исходного продукта. Данные по термическим превращениям 1,1,1-дифторхлорэтана на γ-Al2O3 в импульсном и проточных режимах приведены в табл. 34.
Из приведенных данных видно, что осуществление процесса в проточном режиме, то есть в тех условиях, когда разделение продуктов реакции не происходит, характеризуется повышенным образованием 1,1,1-трифторэтана – продукта гидрофторирования 1,1-дифторэтилена; этот результат свидетельствует о возможности каталитического взаимодействия целевых галоидолефинов, образующихся при дехлорировании фторхлоруглеродов водородом, с фтористым и хлористым водородом.
В дальнейшем, для оценки дегидрогалогенирующей, а по принципу детального равновесия и гидрогалогенирующей активности катализаторов, нами была изучена модельная реакция дегидрохлорирования хлористого этила. На рис. 8 приведены данные, показывающие, что интенсивность образования побочных продуктов при каталитическом дехлорировании фторхлоруглеродов, в частности 1,2-дихлоргексафторпропана, коррелирует с активностью этих же катализаторов в дегидрохлорировании C2H5Cl.
Таблица 34. Термическое превращение 1,1,1-дифторхлорэтана на γ-Al2O3 (скорость подачи субстрата – 7200 час-1).
|
# оп. |
Режим |
Температура, К |
Степень конверсии, % |
Состав газообразных продуктов реакции, объемн. % |
||
|
СН2=СF2 |
СН3-СF3 |
СН2=СFCl |
||||
|
1 |
Импульсный |
723 |
>99 |
26,3 |
4,1 |
69,6 |
|
2 |
То же |
773 |
>99 |
30,7 |
1,9 |
67,4 |
|
3 |
Проточный |
723 |
96 |
20,3 |
44,7 |
35,0 |
|
4 |
То же |
773 |
98 |
30,0 |
29,6 |
40,4 |
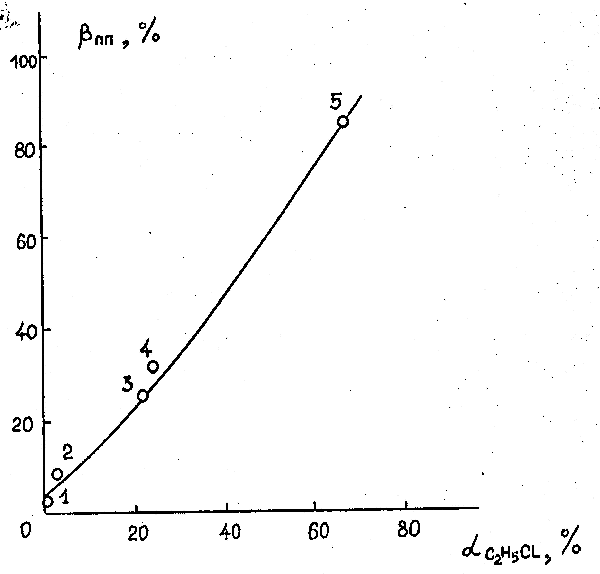
Рисунок 8. Корреляция степени конверсии (673 К) хлористого этила (α C2H5Cl) и выхода побочных продуктов (743 К) при дехлорировании 1,2-дихлоргексафторпропана водородом (β пп) на катализаторах: 1 – Ni/α-AI2O3; 2 – α-AI2O3; 3 – AlF3; 4 – Ni/γ-Al2O3 и 5 – γ-Al2O3.
Этот результат свидетельствует о протекании побочных реакций при дехлорировании фторхлоруглеродов водородом на кислотных, электрофильных центрах катализаторов, обуславливающих их активность в модельной реакции дегидрохлорирования хлористого этила.
При обсуждении результатов каталитического дехлорирования 1,2-дифтортетрахлорэтана отмечалось повышенное образование 1,2-дифторэтилена на Ni /γ-Al2O3. Учитывая высокую активность никеля в гидрировании водородом, а также повышенную электрофильную активность носителя – γ-Al2O3 , можно, по аналогии, предложить следующий механизм внедрения водорода в молекулу галоидолефина:
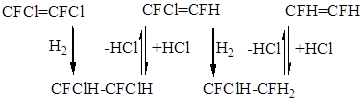
Выше отмечалось, что фторхлоролефины обладают неодинаковым сродством к хлору; это обстоятельство создает термодинамические предпосылки для взаимодействия целевых галоидолефинов с продуктами диспропорционирования исходного субстрата; такое взаимодействие также приведет к уменьшению селективности процесса. О возможности такого типа реакции свидетельствуют результаты прямых экспериментов по контактированию галоидолефинов и фторхлоруглеродов (табл. 35).
Проведенное исследование, таким образом, позволило выявить общие для всех субстратов типы побочных реакций (диспропорционирования, гидро-дегидрогалогенирования, присоединения водорода по двойной связи, дехлорирования галоидлефинами) и установить связь интенсивности их протекания и природы активных центров катализатора.
Таблица 35. Дехлорирование фторхлоруглеродов галоидолефинами (катализатор – активированный уголь, скорость подачи эквимолярной смеси реагентов – 180 час-1).
|
Реакция |
Температура, К |
Степень конверсии фторхлоруглерода, % |
Суммарный выход продуктов реакции, % |
|
|
1.C2F4+C2F3Cl3=C2F4Cl2+C2F3Cl |
593 |
20,6 |
94,9 |
|
|
693 |
57,1 |
93,7 |
||
|
793 |
78,9 |
70,6 |
||
|
2.C2F4+C2F2Cl4=C2F4Cl2+C2F2Cl2 |
593 |
90,3 |
85,0 |
|
|
693 |
95,7 |
94,5 |
||
|
3.C2F3Cl+C2F2Cl4=C2F3Cl3+C2F2Cl2 |
593 |
71,8 |
61,3 |
|
В заключение отметим, что эффективность катализаторов дехлорирования фторхлоруглеродов водородом (активность, селективность, стабильность) во многом определяется природой каталитического носителя; использование α-AI2O3 позволило подобрать на его основе эффективные агрессивостойкие катализаторы и разработать технологию синтеза гексафторпропилена и 1,2-дифтордихлорэтилена селективным дехлорированием соответствующих фторхлоруглеродов.
окончание в следующем выпуске
Список литературы
2. Теддер Д.М. Фторирование органических соединений элементарным фтором. В кн."Успехи химии фтора", т. I-II -М.-Л. : Химия, 1964, с. 380-423.
3. Оркин В.Л., Чайкин A.M. Определение констант скорости образования атомов в реакциях молекулярного фтора с окисью азота, этиленом и тетрафторэтиленом. Кинетикаикатализ, 1982,т.23,в.З,с.529-533.
4. Anson P.O., Fredricks P.S., Tedder J.M. Free-radical substitutionin aliphatic compounds. Part 1. Halogenation of n-butahe and isobutane in the gas phase. J.Chem.Soc, 1959, March, p.918-922.
5. Fredricks P.S., Tedder. J.M. Free-radical substitution in aliphatic compounds. Part 11. Halogenation of the n-butylhalides. -J.Chem.Soc, 1960, Jan., p. 144-150.
6. Purington S.T., Kagen B.S., Patric T.B. The application of elemental fluorine in organic synthesis. Chem.Rev., 1986, № 86, p.997-1018.
7. Aikman R.E., Lagow E.J. Syhthesis of tetra-cis-(perfluorocyclohexyl)-methane and bis-(perfluorocyclohexyl)-methane by direct fluorination. -J.Org.Chem., 1982, v.47, № 14, p.2789-2790.
8. Еременко Л.Т., Орешко Г.В. Фторирование элементарным фтором лабильных полинитросоединений – аддуктов реакции Михаэля. -Изв. АН СССР, сер. Химия, 1969, № 2, с.479.
9. Еременко Л.Т., Нацибуллин Ф.Я., Боровинская Й.П., Карпова Н.Д. Синтез перфторнитроэтанов. -Изв. АН СССР. Сер.хим., 1968, № 2, с.429-430.
10. Патент США 4523039. Способ получения перфорированных простых эфиров. /Лагов Р.Дж., Герхарт Дж.Е. -Опубл. 11.06.85, РЖ Химия, 1986, 7Н36.
11. Патент США 3242218. Способ получения фторуглеродных полиэфиров. /Миллер В.Т. -Опубл.22.03.66,РЖХимия, 1967, 12C232.
12. Des Martean D.D, Fluoroperoxytrifluoromethane CF300F. Preparation from trifluoromethyl hydroperoxide and fluorine in the presence of cesium fluoride. -Inorg Chem., 1972, v.11, № 1, p.193-195.
13. Merritt R.F., Johnson F.A. Direct fluorination. Addition of fluorine to indenes and.acenaphthylenes, -J.Org.Chem., 1966, v.31. № 6, p.1859-1863.
14. Merritt R.F, Johnson F.A. Direct fluorination of steroidal olefines to cis-vicinal difluorides. –J.Am.Chem.Soc., 1966, v.88, № 8, p.1822-1823.
15. Патент США.3487093. Фторированные олефины. Меррит Р.Ф. –Опубл.30.12.69,РЖХимия, 1970, 23Н29.
16. Grakauskas V. Direct liquid phase fluorination of halogenated aromatic compounds. -J.Org.Chem., 1969, v.34, № 10, p.2835-39.
17. Brooke G.M., Chambers B.D., Heyes J., Musgrave W.K.R. Direct preparation and some reactions ofсhlorofluorobenzenes. -J.Chem.Soc., 1964, Febr., p.729-735.
18. Merritt R.F, The polar addition of molecular fluorine to acetylenes. –J.Org.Chem., 1967, v.32, № 12, p.4124-4126.
19. Шеппард У., Шартс К. Органическая химия фтора. -
М.: Мир, 1972, с.52, 89, 112.
20. Миллер В., Эренфельд Дж, Фелан Дж., Пробер М., Рид Ш, Фторирование полностью галоидированных олефинов.«Химияфтора», № 2, -М.:ИЛ, 1950,с.228-240.
21. Miller W.T. My early days in fluorine chemistry. –J.Fluor.Chem., 1981, v.18, p.305-321.
22. Miller W.T., Stoffer J.O., Fuller G., Currie A.C. The mechanism of fluorination. IV. The effect of temperature and of fluorine concentration on reaction. A new fluorination apparatus. –
J.Am.Chem.Soc., 1964, v.86, № 1, p.51–56.
23. Исикава Нобуо, Китацумэ Томоя. Новые методы фторирования ароматических соединений. –Юкки госэй качаку кёкайси., 1976, т.34, № 3, с.173-178, РЖ Химия, 1976, 23Ж360.
24. Патент Японии 55-18695. Способ фторирования./ Маруо Кэйити, Мисаки Сусуму. –Опубл. 21.05.80, РЖ Химия, 1981, 5
Н122.
25. Sirip L.A., Lagow R.J. Direct fluorination of 2,2,4,4-tetramethyl pentane. Sterically protected residual protons. –J.Org.Chem., 1977, v.42, № 21, p.3437-3438.
26. Kowanko N., Branthaver J.F., Sugihara J.M. Direct liquid-phase fluorination of petroleus. –Fuel, 1978, v.57, № 12, p.769-775.
27. Патент США 40004996. Фторирование органических соединений./ Коллониц Дж. –Опубл. 25.01.77, РЖ Химия, 1977, 23Н57.
28. Патент Великобритании 1077065. Фторирование нитросоединений./ Гракаускас В., Хэмел Э.Э. –Опубл. 26.07.67, РЖ Химия, 1975, 16Н84.
29. Merritt R.F. Direct fluorination of 1,1-diphenylethylene. –J.Org.Chem., 1966, v.31, № 11, p.3871-3873.
30. Merritt R.F. The polar fluorination of propenylbenzene. –J.Am.Chem.Soc., 1967, v.89, № 3, p.609-612.
31. Merritt R.F., Johnson F.A. Low-temperature fluorination of Schiff bases. -J.Org.Chem., 1967, v.32, № 2, p.416-419.
32. Misaki S. Direct fluorination of phenol and cresols. –
J.Fluor.Chem., 1981, v.17, № 2, p.159-171.
33. Maxwell A.F., Detoro F.E., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The jet fluorination of certain aliphatic hydrocarbons as oriented and controlled by operation conditions. -J.Am.Chem.Soc., 1960, v.82, № 22, p.5827-5830.
34. Attaway J.A., Groth R.H., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The fluorination of some amides, nitriles and of methylthiocyanate. -J.Am.Chem.Soc., 1959, v.81, № 14, p.3599-3603.
35. Robson P., Mc Longhlin V.C.R., Hynes J.B., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIV. The jet fluorination of hydrogen cyanide, cyanogen, methylamine and ethylenediamine. Pyrolysis and fluorinolysis of selected products. -J.Am.Chem.Soc., 1961, v.83, № 24, p.5010-5015.
36. Boffenberg K. Substitution von benzol durch elementares Fluor in der Gasphase. –Chem.Ztg.,1972, v.96, № 2, p.84-92.
37. Патент Японии 58-41829. Получение октафторпропана./ Фукуи Сиро, Йонэда Хадзимэ. –Опубл.11.03.83,РЖХимия, 1984, 5Н11.
38. Hayes L.J., Dixon D.D. Direct fluorination of polyester and related compounds. –J.Fluor.Chem., 1977, v.10, № 1, p.1-16.
39. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropolyethers by direct fluorination: a novel preparation for perfluoro (polypropylene oxide) ethers and perfluoro (polymethylene oxide) ethers. –J.Chem.Soc., 1981, part 1, № 5, p.1321-1328.
40. Патент США 4113772. Метод получения олигомеров перфторэфиров с концевыми карбоксильными группами. / Лагов Р.Дж. –Опубл.12.09.78,РЖХимия, 1979, 15Н14.
41. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropoly (ethylene glycol) ethers by direct fluorination. –J.Org.Chem., 1978, v.43, № 23, p.4505-4509.
42. Adcock J.L., Znoue Shoji, Lagow R.J. Simultaneous fluorination and functionalization of hydrocarbon polymers. –J.Amer.Chem., 1978, v.100, № 6, p.1948-1950.
43. Lagow R.J., Margrave J.L. The controlled reaction of hydrocarbopolymers with elemental fluorine. –J.Polym.Sci.: Polym.Lett.Ed., 1974, v.12, № 4, p.177-184.
44. Pat. 3775489 (USA). Process for fluorination of aromatic and polynuclear hydrocarbon compounds and fluorocarbons produced thereby. Margrave J.L., Lagow R.J. –Publ. 27.11.73.
45. Lagow R.J., Maraschin N.J. Direct fluorination of cyclic and bicyclic hydrocarbons. –7th Int.Symp.Fluorine Chem., Santa Cruz, Calif., 1973, s.l., s.a., p.1-25.
46. Gerhardt G.E., Dumitru E.T., Lagow R.J. Synthesis of hightbranched perfluoroethers by direct fluorination, promising new materials based on the hexafluoroacetone –ethylene copolymer. –J.Polym.Sci., 1980, v.18, № 1, p.157-169.
47. Robertson G., Liu E.K.S., Lagow R.J. Synthesis of perfluoroadamantane compounds by direct fluorination. –J.Org.Chem., 1978, v.43, № 26, p.4981-4983.
48. Lagow R.J. Large scale synthesis of organofluorine compounds using elemental fluorine; a third Simons cell. –Int.Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.2.
49. Adcock J.L., Lagow R.J. The synthesis of the fluorinated ethers “perfluoroglyme” and “perfluorodiglyme” by direct fluorination. –J.Org.Chem., 1973, v.38, № 20, p.3617-3618.
50. Koshar R.J., Hausted D.R., Meiklejohn R.A. Organic fluoronitrogens. V. Bis(difluoroamino)difluoromethane. –
J.Org.Chem., 1966, v.31, № 12, p.4232-4234.
51. Koshar R.J., Hausted D.R., Wright C.D. Organic fluoronitrogens. VII. Tris(difluoroamino)fluoromethane and related compounds. –J.Org.Chem., 1967, v.32, № 12, p.3859-3864.
52. Патент США 3981783. Процесс электрохимического фторирования с дополнительной подачей водорода и увеличением выхода по току. / Чайлдс В.В. –Опубл.21.09.76, РЖ Химия, 1977, 14Л237.
53. Патент Германии 2106870. Способ электрохимического фторирования органических соединений./Восс П., Нидерпрум Х., Кауль Г., Трепп Р., -Опубл. 24.02.77, РЖ Химия, 1977, 24Н16.
54. Патент Германии 2302132, 1976. Способ получения разветвлённых перфторалканов. /Беннингер С. -Опубл. 23.12.76, РЖ Химия, 1977, 24Л202.
55. Патент США 4035250. Способ получения перфторгептана. /Вальтерс Х.С., Чайдс В.В. -Опубл.12.07.77, РЖ Химия, 1978, 6Н22.
56. Патент Японии 53-18488. Способ получения перфторциклоалканов. /Сато Дайсукэ, Ямамути Коити, Мурасима Рёширо. -Опубл. 15.06.78, РЖ Химия, 1979, 9Н121.
57. Патент США 3662009. Получение ненасыщенных фторсодержащих соединений. /Хадчинсон В.М. -Опубл. 09.05.72, РЖ Химия, 1973, 9Н12.
58. Гольдштейн Б.В., Серушкин И.Л., Никонорова Н.И. О роли фторида никеля в реакциях электрохимических соединений. –Тр. ГИПХ, № 39, часть 1, инв. № Т-1863., Л., 1975, с.43-47.
59. Фаулер Р.Д., Берфорд Н.Б., Гамильтон Дж.М., Свит Р.Дж., Уэбер К.Е., Каспер Дж.С. Лайтайн Н. Синтез фторуглеродов. В кн. «Химия фтора», сб. № 1 –М.: ИЛ, 1950, с.91-113.
60. Беннер Р.Дж., Беннинг А.Ф., Доунинг Ф.Б., Ирвин С.Ф., Джонсон К.С., Линч А.Л., Пармели Х.М., Уирт У.У. Фторуглероды, полученные фторированием углеводородов трехфтористым кобальтом. В кн. «Химия фтора», сб. № 1 –М.: ИЛ, 1950, с.114-128.
61. Фаулер Р.Д., Андерсон Х.С., Гамильтон Дж.Н., Берфорд Н.Б., Спагетти А., Битердих С.Б., Лайтайн Н. Фториды металлов, применяемые в синтезе фторуглеродов. В кн. «Химия фтора», сб. № 1 –М.:ИЛ, 1950, с.143-153.
62. Патент Японии 60-81134. Получение октафторпропана. /Катамура Коити, Кагэяма Ютака, Накаяма Хидэтоси. –Опубл. 09.05.85, РЖ Химия, 1986, 12Н25.
63. Патент Японии 60-10933. Получение гексафторэтана./ Катамура Коити, Кагэяма Ютака, Накаяма Хидэтоси. –Опубл. 15.06.85, РЖ Химия, 1986, 11Н21.
64.PakV.,PekaJ.,CermakV.,SykoraF.,PetrzilaV.Poloprovoznizarizeniprovyrobuperfluororganickychlatek.
–Chem.Prum., 1978,v.28, № 9,p.467-470.
65. Патент Франции 2381732. Усовершенствованный способ перфторирования циклических углеводородов. / Мур Р.Е. –
Опубл. 27.10.78, Изобретения в СССР и за рубежом, 1979, в.55, № 5, с.67.
66. Патент США 4143079. Способ получения перфтор-1-метил-4-изопропилциклогексана из пинена. / Мур Р.Е. –Опубл. 06.03.79, Изобретения в СССР и за рубежом, 1979, в.55, No 21, с.20.
67. Патент Великобритании 1597914. Способ перфторирования циклических углеводородов. / Сантэч инкорп. –Опубл. 16.09.81, Изобретения в СССР и за рубежом, 1982, в.57, № 12, с.64.
68. Burdon J. The exhaustive fluorination of aliphatic compounds.
–Int. Symp. “Cent. ...”, Paris, 1936, p.9.
69. Moore R. E., Driscoll G. Perfluorination of bicyclic and tricyclic hydrocarbons. –IV-th Winter Fluorine Conf., Abstr, Daytona Bearen, 1979, p.8.
70. Асович В.С., Прокудин И.П. Особенности фторирования органических веществ четырехфтористым церием. –Тр. ГИПХ, № 40, инв. № Т-2117, Л., 1976, с.25-31.
71. Исикава Н., Кобаяси Ё. Фтор. Химия и применение. –
М.: Мир, 1982, с.91.
72. Кэйджи Дж..Х., Гроссе А.В., Барбер Е.Дж., Бэрджэр Л.Л., Шелдон З.Д. Получение фторуглеродов каталитическим фторированием углеводородов. В кн. «Химия фтора», сб. № 1 –М.: ИЛ., 1950, с.129-135.
73. Бигелоу Л.А. Действие элементарного фтора на органические соединения. В кн. «Фтор и его соединения (под ред. Дж. Саймсона), т.1 –М.: ИЛ., 1953, с.314-335.
74. Musgrave W. K. R., Smith F. Organic Fluorides. PartІІ. The effect of metals on the fluorunationof hydrocarbons. –J. Chem. Soc., 1949, p.3026-3028.
75. Брик Т. Дж. Фторуглероды; их свойства и производство во время войны. В кн. «Фтор и его соединения» (под ред. СаймонсаДж.),т.І –М.:ИЛ, 1953,с.355-388.
76. Maraschin N. J., Catsikis B. D., Davis L. H., Larvinen J., Lagov R. J. Synthesis of structurally unusual fluorocarbons by direct fluorocarbon. –J.Am. Chem. Soc., 1975, v. 97, № 3, p.513-517.
77. Патент США 4113453. Аппарат для низкотемпературного прямого фторирования. / Лагов Р.Дж, Адкок И.Л., Марашин Н.И. –Опубл. 12.09.78, РЖ Химия, 1979, 19И130.
78. Патент США 4281119. Аппарат для прямого фторирования с контролируемым охлаждением по зонам. / Лагов Р.Дж., Адкок И.Л., Марашин Н.И. –Опубл. 28.07.81, РЖ Химия, 1982, 9Н225.
79. Рахимов А.И., Химия и технология фторорганических соединений. –М.: Химия, 1986, с.9-10.
80. Schmeisser M., Ehlers K. P., Sartori P. Dichlorhexafluorpropan durch fluorierung von 1,2-dichlorpropan. –Angew. Chem., 1967, v.79,№ 13, p.622.
81. Margrave W. K. R., Smith F. Organic Fluorides. PartІ. Fluorination of hydrocarbons. –J. Chem. Sоc., November, 1949, p.3021-3026.
82. Гроссе А.В., Кэйди Дж. Х. Свойства фторуглеродов. В кн. «Химия фтора: сб. № 1 –М.: ИЛ, 1950, с.35-64.
83. Patent GB 1281822. Improved Fluorination Process. / Kingdom R. J., Bond G. D. –Publ. 19.07.72.
84. Hill M. Process and market development of fluorocarbon fluids. –Chem.аnd Ind., 1975, № 3, p.118-121.
85. Патент Франции 2028457 (В). Способ фторирования. / Империал Смелтинг Корп. –Опубл. 15.01.70, Офф. Бюлл. Франции, Химия и металлургия, 1970, № 45-48, ч. І, с.44.
86. Патент США 4220606. Способ получения перфторпроизводных из цикличеких углеводородов. / Мурр Р.Е. –Опубл.02.09.80,РЖХимия, 1981, 9Н105.
87. Patent US 3480667. Method of producing fluorinated compounds. / Siegart W. R., Blackley W. D. –Publ. 25.11.69.
88. Патент США 4330475. Аэрозольный способ прямого фторирования и устройство для этой цели. / Адкок Д.Л., Рэнк Е.Б. –Опубл.18.05.82,РЖХимия, 1983,ІІН76.
89. Ruff J. K. The catalytic fluorination of perfluorocarbon nitriles and imines. –J. Org. Chem., 1967, v.32,№ 5, p.1675-1677.
90. Lusting M., Ruff J. K. Fluorination of some perfluoro alkyliminosulfurdifluorides. –Inorg. Chem., 1965,v.4, № 10,p.1444-1446.
91. Фокин А.В., Столяров В.П., Радченко В.П. Синтезполифторамино-и α-фторнитросоединений на основе реакций газообразного фтора. –Изв. АН СССР, сер. хим., 1982, № 8, с.1853-1861.
92. Фокин А.В., Галахов В.С., Узун А.Т. и др. Реакция фтора с солями щелочных металлов динитроацетонитрила в присутствии фторидов калия или кальция. –Изв. АН СССР, сер. хим., 1974, № 2, с.456-458.
93. Фокин А.В., Узун А.Т., Столяров В.П. Бис (дифторамино) фторацетальдегид –новый представитель α, α-бис (дифторамино) альдегидов. –Изв. АН СССР, Сер. хим., 1982, № 6, с.1438.
94. Еременко Л.Т., Нацибуллин Ф.Я., Боровинская И.П., Карпова Н.Д. Синтез перфторнитроэтанов. –Изв. АНСССР,Сер.хим., 1968, № 3,с.431-432.
95. Sekiya A., Des Martean D. D. Synthesis of 1,1-bis (fluoroxy) –perhaloalkanes by reaction of fluorinated carboxylic acids with fluorine in the presence of cesium fluoride. –Inorg. Chem., 1980, v. 19, № 5, p.1328-1330.
96. Lu S., Des Martean D. D. Direct synthesis of fluorinated peroxides. 7. Perfluoro-tret-butyl-fluoroformyl peroxide. –Inorg. Chem., 1978, v. 17, № 2, p.304-306.
97. Schack C. J. A new synthesis of difluoraminotrifluoromethane. –J. Fluor. Chem., 1981,v. 18,no 4,p.583-586.
98. Патент Германии 2712732. Способ получения октафторпропана. /Халац С.П. –Опубл. 28.09.75, РЖ Химия, 1979, 14Н14.
99. Патент США 4158023. Способ получения октафторпропана. / Халац С.П. –Опубл. 12.06.79. Изобретения в СССР и за рубежом, 1979, в. 55, № 24, с.121.
100. Патент Великобритании 1568020.Способ получения октафторпропана. / Халац С.П. –Опубл. 21.05.80. Изобретения в СССР и за рубежом, 1981, в. 55, № 3, с.54.
101. Патент США 4377715. Получение перфторпропана. /Нучка Х.Р., Хино И.Б., Эйбэк Р.Е., Робинсон Н.А. –Опубл. 22.03.83, Изобретения в СССР и за рубежом, 1983, в.57, № 23, с.88.
102. Европейский патент 0031519. Способ фторирования органических соединений элементарным фтором. / Эллайд Кэмикэл Ко. –Опубл. 08.07.81, Изобретения в СССР и за рубежом, 1983, в.57, No 1, с.20.
103. Европейский патент 0032210. Способ фторирования органических соединений фтором в трубчатом реакторе из пористого металла в присутствии перфторированного разбавителя. / Эллайд Кэмикл Ко. –Опубл. 22.07.81, Изобретения в СССР и за рубежом, 1983, в.57, № 2, с.27.
104. Патент США 4513154. Способ проведения последовательно-конкурирующих газофазных реакций. / Курц Б.Е. –Опубл. 23.04.85, РЖ Химия, 1986, 4Н25.
105. Патент США 2831035. Производство фторированных углеродов. / Тучковский Э.А., Вульф К. –Опубл. 15.04.58, РЖ Химия, 1960, 85740.
106. Патент США 3709800. Получение перфторированных углеводородов./ Фокс Х.М. –Опубл. 09.01.73, РЖ Химия, 1973, 22Н23.
107. Патент Японии 5608. Фторсодержащие галоидуглеводороды. / Осиба Такаси. –Опубл. 22.03.65, РЖ Химия, 1968, 5Н35.
108. Патент США 2681267. Процесс улучшения каталитических свойств фтористого алюминия и продуктов из него. / Калфи Дж. Д., Миллер Ч.Б. –Опубл.15.06.54,РЖХимия, 1956, № 14, 45553.
109. Hohorst F. A., Shreeve J. M. Bis-(fluoroxy)-difluoromethane,CF2(OF)2. –J. Am. Chem. Soc., 1967, v.89, № 8, p.1809-1819.
110. Walker N., Des Martean D. D. Direct Synthesis of fluorocarbon peroxides.ІІІ. The addition of chloroperoxytrifluoromethane to olefins. –J. Am. Chem.Soc., 1975, v.97,№ 1, p.13-17.
111. Патент США 4499024. Непрерывный способ получения бис (фторокси) дифторметана. / Филолт М.Ж. –Опубл.12.02.85,РЖХимия, 1985, 20Н14.
112. Lusting M. A., Pitochelli A. R., Ruff J. K. The catalytic addition of fluorine to a carbonyl group. Preparation of fluoroxy compounds. –J. Am. Chem. Soc., 1967, v. 89, № 12, p.2841-2843.
113. Ruff J. K., Pitochelli A. R., Lusting M. A. A simple synthesis of fluoroxyperfluoroalkyl compounds. -J. Am. Chem. Soc., 1966, v.88,№ 19, p.4531-4532.
114. Kennedy R. C., Cady G. H. Reaction of carbonyl fluoride with fluorine in the presence of various fluorides as catalysts. –J. Fluor. Chem., 1973,v.3, № 1, p.141-154.
115. Мухаметшин Ф. М. Успехи химии фторорганических гипогалогенитов и родственных соединений. –Успехи химии, 1980, т. 49, № 7, с.1260-1288.
116. Мухаметшин Ф.М. Гипофториты и их применение в органическом синтезе. –В кн. «Новые фторирующие реагенты в органическом синтезе». –Новосибирск: Наука, 1987, с.140-196.
117. Cady G. H. Proposed mechanisms of catalysis of fluorination of CF2O. –Anales. Asoc. Quim. Argentina, 1971, v.59, p.3-4, p.125-131.
118. Patent US 3230264. Reaction of carbonyl fluoride with fluorine. –Roger S. P., Cady G.H. –Publ. 18.01.66.
119. Wechsberg M., Cady G.H. Comparative studies of catalytic fluorination of carbon monoxide with elementary fluorine. –J. Am. Chem. Soc., 1969, v.91, p.4432-4435.
120. Kellog K. B., Cady G.H. Trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1948, v.70,№ 12, p.3986-3988.
121. Gervasi J.A., Brown M., Bigelow L. A. The action of elemental fluorine upon organic compounds. XX. The fluorination of mono-, di-and trimethylamine, ethylenediamine and ethyleneimine. –J. Am. Chem. Soc., 1956, v.78, № 8, p.1679-1682.
122. Avonda F. P., Gervasi J. A., Bigelow L.A. The action of elemental fluorine upon organic compounds. XXІ. The fluorination of malononitrile and dimethylformamide. –J. Am. Chem. Soc., 1956, v.78,№ 12, p.2798-2800.
123. Тойтельбойм М.А., Шойхет А.А., Каплунов М.Г., Веденеев В.И. Энергетические разветвления цепей в реакциях трифторметилгипофторита с галоидметанами. –Кинетика и катализ, 1981, т. 22, в.2, с.298.
124. Тойтельбойм М.А., Шойхет А.А., Веденеев В.И. Энергетические разветвления цепей в реакциях трифторметилгипофторита с галоидметанами. 2. Механизм отрицательного взаимодействия цепей. –Кинетика и катализ,
1981, т.22, в.3, с.564-568.
125. Веденеев В.И., Парийская А.В. Механизм фторирования метана и его фторпроизводных. І. Сравнение скоростей фторирования метана, фторметана, дифторметана и трифторметана. –Кинетика и катализ, 1971, т.12, в.1, с.21-26.
126. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его производных. 2. Дифторметан –Кинетика и катализ, 1971, т.12, в.2, с.293-298.
127. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его фторпроизводных. 3. Фтористый метил. –
Кинетика и катализ, 1971, т.12, в.3, с.543-548.
128. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его фторпроизводных. 4. Метан. –Кинетика и атализ, 1971, т. 12, в.4, с. 839-842.
129. Надточенко В.А., Федотов Н.Б., Веденеев В.И., Саркисов О.М. О реакции колебательно возбужденной молекулы СН3F с фтором. –Докл. АН СССР, 1978, т.238, № 6, с.1391-1394.
130. Парийская А.В., Веденеев В.И. О природе задержек самовоспламенения в системе СН3F + F2 + O2 + Не. –Кинетика и катализ, т.14, в.6, с.1365-1369.
131. Веденеев В.И., Тейтельбойм М.А., Шойхет А.А. Фотохимическое фторирование фтороформа в присутствии кислорода. –Изв. АН СССР, 1976, № 9, с.1968-1970.
132. Медведев Б.А., Тейтельбойм М.А., Шилов А.Е. Химическая активация солекулы СHFCl2 в реакции СНCl2+ F2→ СНСl2F + F . –Кинетика и катализ, т.12, в.2, с.269-275.
133. Медведев Б.А., Тейтельбойм М.А., Шилов А.Е. Механизм газофазной реакции хлорфторметана с молекулярным фтором. –Кинетика и катализ, 1971, т.12, в.3, с.49-751.
134. Веденеев В.И., Медведев Б.А., Тейтельбойм М.А. Газофазное фторирование дифторметана при повышенных давлениях инертного газа. –Кинетика и катализ, т.13, в.1, с.50-53.
135. Обвивальнева А.А., Федотов В.Г. Возбужденные молекулы в реакции фтора с ацетоном (фторацетоны; синтез; механизм). –Кинетика и катализ, 1981, т.22, в.5, с.1095-1099.
136. Капралова Г.А., Марголина Е.М., Русин Л.Ю., Чайкин А.М., Шилов А.Е. Роль колебательно-возбужденных молекул в реакциях фторирования молекулярным фтором. –В сб. «5 Междунар. симпозиум по химии фтора», Тезисы докл., М., : Наука, 1969, с.102-104.
137. Кнунянц И.Л, Гамбарян Н.П., Рохлин Е.М. Карбены. (Соединения двухвалентного углерода, промежуточно образующиеся в органических реакциях). –Успехи химии, 1958, т.27, в.12, с.1361-1436.
138. Мухаметшин Ф.М., Жирнов О.М., Айнагос Н.А., Петров Ю.И., Ляпунов М.И. О некоторых особенностях гипофторитного метода получения перфторметилвинилового эфира. Сообщ. 2. Синтез трифторметилгипофторита. –Тр. ГИПХ, № 69, инв. № Т-2769, Л., 1980, с.73-78.
139. Мухаметшин Ф.М., Поврозник С.В., Жирнов О.М. Исследование кинетики и механизма каталитической реакции образования трифторметилгипофторита. –Тр. ГИПХ, № 103, инв. № Т-2981, Л., 1983, с.75-80.
140. Авторское свидетельство СССР 1141707. Способ получения 1,1-дифтор-1-хлорэтана или 1,1,2,2-тетрафтордихлорэтана. /Захаров В.Ю., Голдинов А.Л., Боровнев Л. М., Голубев А.Н., Калашникова Н.А., Захарова О.М., Любимова Л.А.
141. Капралова Г.А., Бубен С.Н., Чайкин А.М. Кинетика и механизм реакции фторирования окиси углерода. –Кинетика и катализ, 1975, т.16, в.3, с.591-595.
142. Поврозник С.В. Разработка способа и технологии получения фторокситрифторметана реакциями элементарного фтора с карбонилсодержащими соединениями. Дисс. канд. техн. наук, 1987, ПФ НПО ГИПХ, 161с.
143. Краснов К.С. Молекулы и химическая связь. –М.: Высш. школа, 1977, с.117.
144. Безмелицын В.Н., Легасов В.А., Чайванов Б.Б. Синтез дифторида криптона с применением термической генерации потоков атомарного фтора. –Докл. АН СССР. Сер. хим., 1977, т.235, № 1, с.96-98.
145. Безмелицын В.Н., Легасов В.А., Спирин С.Н., Чайванов Б.Б. Кинетика каталитической атомизации фтора. –Докл. АН СССР. Сер.физ.химия, 1982, т.262, № 5, с.1153-1157.
146. Палкина Л.А., Спирин С.Н., Тищенко П.П. процессы с участием атомарного фтора. –Докл. АН СССР. Сер.физ.химия, 1982, т.262, с.1428-1433.
147. Безмелицын В.Н., Васильев А.А., Синянский В.Ф., Чайванов Б.Б. Кинетика термокаталитической диссоциации фтора на поверхности никеля. –
Тезисы докладов на 8 Всесоюзн. симп. по химии неорг. фторидов, 1987, М.: Наука, с.59.
148. Никоноров Ю.И. Фториды никеля. Их каталитическая и окислительная активность. –Тезисы докладов на 5 Всесоюзн. симп. по химии неорг. фторидов, 1978, М.: Наука, с.205.
149. Рысс И.Г. Химия фтора и его неорганических соединений. –М.: Госхимиздат, 1956, с.576-579.
150. Redwood M.E., Willis C. J. Fully fluorinated alkoxides. PartІ. Trifluoromethoxides of alkali metals. –Canad. J.Chem., 1965,v.43,p.1893-1898.
151. Губанов В.А., Веретенников Н.В., Тройчанская П.Е., Долгопольский И.М. Синтез перфторалкоксидов металлов І группы и термографическое изучение их свойств. –Ж. Орг. хим., 1975, т.ІІ, № 2, с.322-325.
152. Levy J. B., Kennedy R. C. Bis (trifluoromethyl) peroxide.І. Thermodynamics of the equilibrium with carbonyl fluoride and trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1972, v.94, no 10, p.3302-3305.
153.Патент Японии 35888.Способполучениятрифторметилгипофторита. /НагасэСюндзи,АбэТакаси,БабаХадзимэ,ОхираКадзуо. –Опубл.09.09.72, РЖХимия, 1973, 14Н120.
154. Патент США 3687825. Способ получения трифторметилгипофторита. Нагасэ Сюндзи, Абэ Такаси, Баба Надзимэ. –Опубл. 29.08.72, РЖ Химия, 1973, 21Н26.
155. Патент США 2983764. Способ получения фторуглеродных соединений./ Кнак Д.Ф. –Опубл. 9.05.61, РЖ Химия, 1962, 14Л53.
156. Патент Японии 56-16429. Олигомеризация гексафторпропилена в газовой фазе. / Хосака Йоносукэ, Хигасидзука Такэси. –Опубл. 17.02.81, РЖ Химия, 1982, 3Н10.
157. Патент Японии 50-45818. Получение олигомеров гексафторпропилена. Кадзава Масахиро, Комацу Тадааки, Мацуока Кимиаки. –Опубл. 08.06.81, РЖ Химия, 1982, 9НІІ.
158. Патент США 4296265. Способ получения олигомеров гексафторпропилена. / Хосака Йоносукэ, Тозука Такаси. –Опубл. 20.10.81, РЖ Химия, 1982, І3НІІ.
159. Патент Германии 3027229. Способ получения олигомеров гексафторпропилена. / Осака Ю., Тозука Т. –Опубл. 07.02.81, Изобретения в СССР и за рубежом, 1981, В.55, № 12, с.30.
160. Патент Бельгии 2055824А. Способ получения олигомеров гексафторпропилена. /Осака И., Тозука Т. –Опубл. 18.07.80, Сборник рефератов НИОКР, обзоров, переводов и деп. рукописей. ДСП. Химия и химическая технология, М., 1986, № 5, с.14.
161. Патент Германии 2616733. Способ получения ди
-и тримеров гексафторпропилена. / Окава М., Комацу Т., Мацуока К. –Опубл. 20.03.80, Изобретения в СССР и за рубежом, 1980, № 15, с.13.
162. Патент Японии 57-2697. Способ получения олигомеров гексафторпропена. / Нэосу К.К. –Опубл. 18.01.82, Изобретения в СССР и за рубежом, 1982, в.57, № 14, с.113.
163. Scherer K. V., Ono T., Jamanouchi K., FernandezR., Henderson P.B. F-2,4-Dimethyl-3-ethyl-3-pentyl and F-2,4-dimethyl-3-isopropyl-3pentyl: stable tert-perfluoroalkyl radicals prepared by addition of fluorine or trifluoromethyl to a perfluoroalkene. –J. Am. Chem. Soc., 1985, v.107, № 3, p.718-719.
164. Scherer K. V., Fernandez R., Henderson P.B. Long lived perfluoroalkyl radicals: preparation by direct fluorination and alternate routes, properties and rates andmechanisms of disappearance. –Int. Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.1.
165. Патент Германии 2332097. Способ получения перфторнонанов. /Халац С.П. –Опубл.16.01. 75,РЖ Химия, 1975, 18Н25.
166. Патент Германии 2332088. Способ получения перфтор-2-метилпентана./ Халац С.П. –Опубл. 16.07.81, Изобретения в СССР и зарубежом, 1981, в.55, № 23, с.21.
167. Аллаяров С.Р., Баркалов И.М., Гольданский В.И., Кирюхин Д.П. Образование стабильных радикалов из фторорганических соединений. –Изв. АН СССР. Сер. хим., 1983, No 6, с.1225-1228.
168. Аллаяров С.Р., Баркалов И.М., Гольданский В.И., Демидов С.В., Кирюхин Д.П., Михайлов А.И. Стабильные радикалы –растворенные перфторалкилы. –Изв. АН СССР. Сер. хим., 1983, № 6, с.1448-1449.
169. Аллаяров С.Р., Михайлов А.И., Баркалов И.М. Новый стабильный перфторалкильный радикал. –Изв. АН СССР. Сер.хим., 1985, № 7, с.1667-1669.
170. Аллаяров С.Р., Баркалов И.М., Лебедов М.Ю., Логинова Н.Н., Михайлов А.И. Образование стабильных радикалов при радиолизе гексафторпропилена. –Изв. АН СССР. Сер. хим., 1986, № 2, с.462-463.
171. Аллаяров С.Р., Баркалов И.М., Гольданский В.И. Образование долгоживущих радикалов при фторировании жидких углеводородов. – Изв. АН СССР. Сер. хим., 1986, № 10, с.394.
172. Аллаяров С.Р., Сумина И.В., Баркалов И.М., Михайлов А.И. Образование стабильных радикалов при
фотолизе перфтор-2,4-диметил-3-этилпентена-2. –Изв. АН СССР, Сер. хим., 1986, № 10, с.2359-2361.
173. Krusic P. J., Scherer K. V. An electron spin resonance study of long lived fluoroalkyl radicals generated by photolysis of hindered perfluoroalkenes. Int. Symp. “Centenary of the discovery of fluorine”. Abstr., Paris, 1986, p.37.
174. Levy J.B., Kennedy R. C. Homolytic displacement reactions of carbon.І. The fluorine-perfluorocyclobutane reactions. –J.Am.Chem.Soc., 1974, v.96, № 15, p.4791-9795.
175. Гольдинов А.Л., Захаров В.Ю., Байбаков П.Я., Жукова В.А., Новикова М.Д. Новые аспекты прямого фторирования. Каталитическое газофазное фторирование фторолефинов; получение перфторуглеродов. –Отчет предприятия п/я А-1619, 1987г., инв. № 1288, 87с.
176. Матвеев С.А., Прокудин Л.П., Тимофеев Н.В. Использование трифторида кобальта для фторирования органических соединений по двойным связям. –Тр. ГИПХ, 1975, № 31, инв. № Т-1728, с.119-127.
177. Асович В.С., Прокудин И.П. Фторирование ацетилена фторидами кобальта и церия. Тр. ГИПХ, № 55, инв. № Т-2552, Л., 1978, с.21-24.
178. Асович В.С., Прокудин И.П., Максимов Б.Н., Степанов В.П. Изучение кинетических закономерностей фторирования ацетилена трехфтористым кобальтом. –Тр. ГИПХ, № 65, инв. № Т-2723, Л., 1979, с.42-47.
179. Котиков С.В., Прокудин И.П., Асович В.С. Изучение кинетических закономерностей фторирования гексафторпропилена трехфтористым кобальтом. –Тр. ГИПХ, № 101,инв. №Т-2986,Л.,ГИПХ, 1985,с.24-27.
180. Burdon J., Knights J.R., Parsons J. W., Tatlow J. C. The fluorination of ethane and ethene over potassium tetrafluorocobaltate (ІІІ) and cobalt trifluoride. –Tetrahedron, 1976, v.32, p.1041-1043.
181. Гольдинов А.Л., Боровнев Л.М., Широкова Н.С., Ковбасюк О.Г., Тишина В.В. Влияние технологических параметров синтеза и чистоты растворителя на свойства фторопласта-50. –Отчет предприятия п/я А-1619, 1987, инв. № 1231.
182. Патент Германии 2451493. Способ получения перфторированных эфиров./ Халац С.П., Клаг Ф. –Опубл.06.05.76,ИзобретениявСССРизарубежом,1982,в.57, № 22,с.5.
183. Patent US 3665041 (USA). Perfluorinated polyethers and process for their preparation. / Sianesi D., Fontanelli R., -Publ. 23.05.72.
184. Рябинин Н.А., Долубенов А.Е., Шматов Л.Е. Фторполиэфиры как антиадгезивы при прессовании перхлората нитрония. –Тр. ГИПХ, № 101, инв. № Т-2986, Л., 1985, с.83-87.
185. Авторское свидетельство СССР 1332758. Способ очистки гексафторпропилена, тетрафторэтилена и их семей от октафторизобутилена. /Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
186. Гольдинов А.Л., Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Разработка технологии получения 2
-перфторметил-4-оксаперфторнонана на основе перфторизобутилена и 1,1,5-тригидроперфтор пентанола. –Отчет предприятия п/я А-1619, 1987, инв. № 1229, 39с.
187. Авторское свидетельство СССР 1148285. 3-Гидропропилфторсульфат в качестве промежуточного продукта для синтеза 2,2,3,3-тетрафторпропионовой кислоты и способ ее получения. /Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
188. Заявка СССР 3966753. Способ получения 2-гидротетрафторэтилфторсульфата. /Фокин А.В., Рапкин А.И., Татаринов А.С. и др. Пол. реш. от 26.09.86.
189. Авторское свидетельство СССР 1233595. Способ получения ω-гидроперфторкарбоновых кислот (его варианты). / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
190. Технические требования к ПФДМЦГ для использования в радиоэлектронике. М.: ИНЭОС АН СССР. Рег. № 12111-10 ДСП от 18.01.85.
191. Исходные данные для разработки жидкости-диэлектрика. М.: ИНЭОС АН СССР. Прил. к вх. № 155 от 18.02.85.
192. Гольдинов А.Л., Захаров В.Ю., Байбаков П.Я. и др. Разработка технологии получения перфтордиметилциклогексана повышенной чистоты. –Отчет предприятия п/я А-1619, 1986, инв. № 1218, 24с.
193. Гольдинов А.Л., Масляков А.И., Захаров В.Ю. и др. О возможности резкого увеличения выпуска дефицитных фторуглеродных жидкостей и смазок. –Отчет предприятия п/я А-1619, 1986, № 1219, 13с.
194. Временный технологический регламент производства перфтордекалина.1981,инв. № 1115.
195. Furin G.G. Synthetic aspects of the fluorination of organic compounds. (Ed.) M.E. Vol’pin./Harwood Academic
Publishers GmbH: United Kingdom, Sov. Sci. Rev., B. Chem., 1991, V.16, Part 1,р.140.
196. Rozen S., Kol M. Olefin epoxidation using elemental fluorine. / J.Org.Chem., 1990. V.55,р.5155-5159.
197. Patent JP 02-131438, (1990). CO719/08. Preparation of hexafluoroethane. /Shimuzu M.C.A. 1990, V.I 13,97031.
198.PatentJP62-193946, (1989).CO719/08,CO17/04. Способ получения фторсодержащих этанов. /Фарука Я., Коно Х. РЖХ, 1990, 2Н 131.
199. Lagow R.J. Important new developments in direct fluorination technology. /Memorial symposium for Prof. Nobuo Ishikawa, Dec. 9-10, 1991, Nippon Kaiun Club, Japan.
200. Lin W.H., Lagow R.J. Synthesis of perfluorodicyclohexano-crown-6 ether. /J.Chem.Soc., Chem.Commun., 1991,р.13-14.
201. Clark W.D., Lin T.Y., Maleknia S.D., Lagow R.J. Synthesis of perfluorotetralkyl ortocarbonates using elemental fluorine. / J.Org.Chem., 1989. V.54.р.1990-1992.
202. Sievert A.C., Tong W.R., Nappa M.J. Preparation D’analogues fluores de la pheromone d’anthanomus grandis. / J.Fluorine Chem., 1991. V.51.р.397-405.
203. Tonelli C., Tortelli V. Photoinduced fluorination of hexafluoropropene trimers: synthesis of branched perfluoroalkanes. /J.Chem.Soc., Perkin Trans I, 1990, № 1,р.23-26.
204. Patent Application EP 344935, (1989). CO8G65/00. Preparation of prefluoroethers by photo-assisted fluorination. / Nappa M.J., Sievert A. C.A. 1990, V. 112, 216229.
205. Patent US 4960951, (1990). CO743/12. Novel perfluoroalkyl ethers by two-step fluorination of polyols./ Nappa M.J., Sievert A.C. Tong W.R. C.A. 1991, V. 111, 216229.
206. Sievert A.C., Tong W.R., Nappa M.J. Synthesis of perfluorinated ethers by an improved solution phase direct fluorination process. / J.Fluorine Chem. 1991.V. 53, р. 397-417.
207. Мизин Г.Г., Симаков Б.В., Шкультецкая Л.В., Молдавский Д.Д. Исследование способа получения перфторэтилциклогексена. / ЖПХ. 1991. т.64 ,в.6. с.293-1296.
208. Moldavsky D.D., Mizin G.G., Shultezkaya L.V., Simakov B.V., Furin G.G. Synthesis of perfluoroethylcyclohexene, its alkoxy and diakylamino derivates and their electrochemical fluorination. / Abstracts of 10-European Symposium on Fluorine Chemistry, Sept. 20-25, 1992, Padua, Italy. Abstracts.B7;J.FluorineChem., 1992.v.58. р.185.
209. Молдавский Д. Д. Перфторорганические соединения: Получение прямым фторированиемуглеводородов. Дисс. докт. хим. наук, 2002, РНЦ Прикладная химия, 268с.
210. Суворов Б.В., Букейханов Н.Р. Окислительные реакции в органическом синтезе. 1978г. М., Химия, 197с.
211. Семенова Г.А., Лейтес И.Л., Аксельрод Ю.В., Маркина М.И., Сергеев С.П.,Харьковская Е.Н. Каталитические и адсорбционные методы очистки газов от сернистых соединений. –Очистка технологических газов. 1977г, М., Химия, 1977, с. 35-37.
212. Панов Г.И. Закономерности гетерогенно-каталитической активации двухатомных молекул (N2, О2, Н2). Новые каталитические системы активации молекулярного азота. Дисс. докт. хим. наук, 1986г. Новосибирск, 374с.
213. Авторское свидетельство ЧССР 220740. Способ получения тетрахлортетрафторпропана. –Паллета О., П. Свобода, Л. Стефан, Я. Квисала. –Опубл. 15.12.85. РЖ Химия, 1986, ІІН24.
214. Патент Японии 60-116637. Получение фторметана. –Такаяма С., Мэйраку Ф., Такоити А., Ковасаки Х. –Опубл. 24.06.85. РЖ Химия, 1986, 9Н16.
215. Марголис Л.Я. Окисление углеводородов на гетерогенных катализаторах. 1977г. М., Химия, с. 296.
216. Каваев М.М., Запорин А.П., Клещев Н.Ф. Каталитическое окисление аммиака. 1983г. М., Химия, с. 31.
217. Технология катализаторов. 1974г. Л.,Химия,с. 138-140.
218. Patent US 2760997. Process for the production of chlorotrifluoroethylene by passing a mixture of trichlorotrifluoroethane and hydrogen through an unobstructed iron tube. / Rucker J. T., Stormon D. B. –Publ. 28.08.56.
219. Patent US 2615925. Preparation of olefinic compounds. / Bordner C.A. –Publ. 28.10.52.
220. Patent US 2704777. Preparation of halogenated olefines / Clark J.W. –Publ. 22.03.55.
221. Patent GB 698386. Improvement in preparation of halogenated olefines / Clark J. W. –Publ. 14.10.53.
222. Patent DE 1044800. Verfahren zur Herstellung von Chlortrifluorathylen / Clark J. W. –Publ. 27.11.58.
223. Patent US 2685606. Preparation of halogenated olefines / Clark J. W. –Publ. 03.08.54 C.A. 48, 127881 (1954).
224. Patent US 2697124. Dehalogenation of fluorohalocarbons / Russel M.M. –Publ. 14.12.54 C. A. 50, 2650 (1956).
225. Patent US 2864873. Production of 1,1,2-trifluoro-2-chloroethylene / Miller C. B., Smith L. B. –Publ. 16.12.58.
226. Патент США 3043889. Получение фтористого этилена. / Смит Л.Б., Вулф К. –Опубл. 10.07.62. РЖ Химия, 1964, 3Н24.
227. Патент Германии 1186847. Получение 1-хлор-1,2,2-трифторэтилена. / Смит Л.Б., Вульф К. –Опубл. 4.10.65, РЖ Химия, 1966, 13Н26.
228. Патент США 3333011. Получение хлортрифторэтилена./Анелло Л.Г., Вульф К. –Опубл. 25.07.67 РЖ Химия, 1969, 14Н30.
229. Патент Японии 60-185734. Получение трифторхлорэтилена. / Моримото Такэси, Морикава Санэсукэ, Фунаяма Кэйди. –Опубл.21.09.85, 19Н37.
230. Европейский патент 0053657. Способ получения хлортрифторэтилена и трифторэтилена. / Канинхэм У.Д., Плэкорз Р.Е., Смит Э.М. –Опубл. 16.06.82. Изобр. в СССР и з/р., вып. 57, № 15, с. 83.
231. Патент Японии 43-8454. Способ получения трифторэтилена. / Саката Хиросукэ. –Опубл. 02.04.68. РЖХимия, 1969, 3Н36.
232. Патент США 2913400. Катализатор из окиси молибдена для конверсии углеводородов. / Буртон У.П., Лефрэнкойс Ф.А., Риблет Е.У. –Опубл. 17.11.59, РЖХимия, 1961, 9К156.
233. Патент США 3354233. Производство тетрафторэтилена. / Анелло Л.Г., Вульф К. –Опубл. 21.11.67, РЖ Химия, 1969, 9Н32.
234. Patent DE 1270547. Verfahren zur Herstellung von Tetrafluorathylen/ Anello Z. G., Wolf C. –Publ. 30.01.69.
235. Патент США 3505417. Дегалоидирование фторгалогенуглеродов. / Гарднер Л.Е. –Опубл. 07.04.70, РЖ Химия, 1971, 11133.
236. Патент США 3636173. Способ каталитического дегалоидирования./ Гарднер Л.Е. –Опубл. 18.01.72, РЖ Химия, 1972, 21Н21.
237. Патент США 3789016. Катализатор гидродегалоидирования. / Гарднер Л.Е. –Опубл. 29.01.74. РЖ Химия, 1975, 2Н217.
238. Патент США 3636172. Дегалоидирование фторгалоидуглеводородов. / Гарднер Л.Е. –Опубл. 18.01.72, РЖ Химия, 1972, 21Н20.
239. Патент США 2900423. Производство перфторпропилена. / Смит Л.Б. –Опубл. 18.08.59. РЖ Химия, 1961, 7Л44.
240. Патент США 2734090. Получение фтористого винилидена. / Кафи Д.Д., Миллер Ч.Б. –Опубл. 07.02.56, РЖ Химия, 1957, 12, 42313.
241. Патент США 3404180. Получение арбонилфторида. / К.К. Лестер. –Опубл. 1.10.68. –РЖ Химия, 1969, 23Н92.
242. Патент Франции 1460246. Способ получения окиси тетрафторэтилена. / Эдисон. –Опубл. 17.10.66. –РЖ Химия, 1968 ІІНІ24.
243. Соколов Л.Ф. Исследование и разработка технологии процессов окисления фторолефинов. –
Дисс. докт. хим. наук. –Ленинград, ВНИИСК, 1979.
244. Авторское свидетельство СССР 190555. Способ получения фтористого водорода. / Гольдинов А.Л., Романов Е.И., Захаров В.Ю. и др.
245. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Термическое обезвреживание фторорганических отходов в водородо-воздушном пламени на опытно-
промышленной установке. –Отчет предприятия п/я А-
1619, 1982, инв. № 887, 18с.
246. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Катализ и инициирование деструктивного окисления, тетрафторэтилена молекулярным
кислородом. Получение карбонилфторида. –Отчет предприятия п/я А-1619, 1983, инв. № 1110, 21с.
247. Патент ГДР 213342. Способ получения фторсодержащих галоидуглеводородов со средней степенью фторирования. / Л. Ингэбург, К. Рэйнфред, М. Дитэр. –Опубл. 24.12.85. –РЖ Химия, 1986, 21Н17.
248. Патент ГДР 203314. Катализатор получения хлорфторметана в газовой фазе. / И. Лелид, Р. Каден, Д. Мросс. –Опубл. 19.10.83. –РЖ Химия, 1984, 17Н181.
249. Патент США 4504686. Способ получения 1,1,2-трифтор-2-хлорэтилдифторметилового эфира. / С. Таками. –Опубл. 12.03.85. –РЖ Химия, 1986, 8Н9.
250. Патент Японии 55-85564. Получение β-трифторметилхлорпиридинов./ Р.Нишима, К. Рудзикама, И.Йокомити, Я. Цудзии, С. Нисимура. –Опубл. 27.06.80. –РЖ Химия, 1981, 19Н177.
251. Патент США 4139568. Способ получения метилфторида. / В. Даниэль, М. Либбс. –Опубл. 13.02.79. –РЖ Химия, 1979, 18Н18.
252. Патент США 4145368. Получение 1,1,1-трифтор-2,2-дихлорэтана. Р. Свинэй, С. Бернард. –Опубл. 20.03.79. –РЖ Химия, 1979, 20Н20.
253. Патент Франции 1453510. Способ очистки дихлортетрафторэтана. / Корп. Электрохимия. –Опубл. 16.08.66. –РЖ Химия, 1968, 11Н39.
254. Виленчик Я.М., Якурнова Г.И., Остапенко Э.А. Синтез трифторацетилфторида на основе окиси тетрафторэтилена. –Тр. ГИПХ, 1985, № 101, с. 147-149.
255. Патент Японии 58-35978. Способ получения пентафторпропионилфторида. / А. Йонотаси, А. Йосио. –Опубл. 08.05.83.-Изобретения в СССР и за рубежом, 1984, вып. 57, № 8, ч.2, с. 130.
256. Патент Японии 58-38231. Получение пентафторпропионилфторида./ И. Йоносукэ, А. Йосио, С. Хироюки. –Опубл. 05.03.83.-РЖ Химия, 1984, 5Н46.
257. Патент США 3250808. Фторсодержащие простые эфиры. / Е.Ф. Мур, А. Мильян, Г. Элеутерио. –Опубл. 10.05.66. –РЖ Химия, 1967, 17Н82.
258. Патент США 3250807. Фторсодержащие карбоновые кислоты с эфирными группировками. / Ч.Г. Фритц, Е.Ф. Мур. –Опубл. 10.05.66. –РЖ Химия, 1967, 17Н84.
259. Патент США 3242218. Способ получения фторуглеродных полиэфиров. / В.Миллер. –Опубл. 22.03.66. –РЖ Химия, 1967, 12С232.
260. Патент ГДР 78376. Способ полимеризации и сополимеризации окиси гексафторпропилена в присутствии гексафторпропилена. / Г. Лотхар. –Опубл. 12.12.70. –РЖ Химия, 1971, 21 С. 361.
261. Патент США 3125599. Полимеры окисей фторуглеродных соединений. / Д. Ворнелл. –Опубл.17.03.64. –РЖХимия, 1965, 15С. 277.
262. Patent US 3291843. Fluorinated vinilethers. / Fritz C.G., Selman S., -Publ. 13.12.66.C.A. 1967, 66, 37427 h.
263. Патент США 4377717. Способ получения перфтор-2-метилпентена-2. / Л.Анелло, Р. Свинэй. –Опубл. 22.03.83. –РЖ Химия, 1983, 24Н1311.
264. Кнунянц И.Л., Фокин А.В., Чебурков Ю.А. Четвертый международный симпозиум по химии фтора в Истес-
Парк (Колорадо, США) –ЖВХО им. Д.И.Менделеева, т. 13, № 3, с. 311-321.
265. Патент США 3449389. Первичные фторзамещенные алкоголяты. / Д. Ворнелл. –Опубл. 10.06.69. –РЖ Химия, 1970, 18Н39.
266. Патент США 3322826. Полимеризация гексафторпропиленэпоксида./ Е.Ф.Мур –Опубл. 0.06.67. –РЖ Химия, 1968, 18С 296.
267. Патент Японии 57-45132. Получение перфтор-2-метил-3-оксагексаноилфторида. / Я. Масаки, М. Сэйдзи. –Опубл. 13.03.82. –РЖ Химия, 1983, 5Н70.
268. Патент США 3274293. Фторсодержащие простые эфиры. / С.Стенлей. –Опубл. 20.09.66. –РЖ Химия, 1968, 5Н161.
269. Патент Германии 2756919. Способ демеризации окиси гексафторпропилена. / Г. Кюхнэ, Ф. Геллер. –Опубл. 05.07.79. –РЖ Химия, 1980, 14Н22.
270. Патент Германии 2924385. Способ димеризации окиси гексафторпропилена. / Г. Кюхнэ. –Опубл. 07.08.80. –РЖ Химия, 1981, 23Н36.
271. Кинле Х., Бадер Э. Активные угли и их промышленное применение. –Л., Химия, 1984, с. 185-192.
272. Патент США 3321515. Способ получения фторированных карбонильных соединений. / Ф.Мур, С.Мильян. –Опубл. 23.05.67.-РЖ Химия, 1968, 15Н48.
273. Беккер Р.А., Асратян Г.В., Дяткин Б.Л. О роли пространственных факторов в реакциях полифторированных α-окисей с аминами. –ЖОрХ, 1973, т. 9, вып. 8, с. 1644-1648.
274. Кнунянц И.Л., Шокина В.В., Тюленева, В.В., Чебурков Ю.А., Аронов Ю.Е. Производные перфторизомасляной кислоты. –Изв. АНСССР,сер.хим., 1966, № 10,с. 1831-1833.
275. Sianesi D., Passety A., Tarli F., The Chemistry of hexafluoropropene epoxide. –J. Org. Chem., 1966,v. 31, № 7,p. 2312-2316.
276. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Инициирование процесса сополимеризации тетрафторэтилена и гексафторпропилена перфторированными диацильными перекисями, синтезированными на основе окиси гексафторпропилена. –Отчет предприятия п/я А-
1619, 1985, инв. № 1154, 28с.
277. Ашмор. Катализ и инициирование химических реакций. М.: Мир, 1966, с. 62.
278. Патент США 3213134. Дециклизация фторированных циклических эфиров. / Д. Морин. –Опубл. 19.10.65. –РЖ Химия, 1967, 9Н60.
279. Патент США 4302608. Способ изомеризации окиси гексафторпропилена до гексафторацетона. / Э.Скури, Д. Милз. –Опубл. 24.11.81. –РЖ Химия, 1982, 20Н56.
280. Патент Японии 58-62131. Получение гексафторацетона. / О. Йосио, А. Йосимаса, М. Макисукэ. –Опубл. 13.04.83. –РЖ Химия, 1984, 12Н30.
281. Патент Японии 58-62130. Получение гексафторацетона. / А. Йосимаса, М. Макисукэ, С. Юкио. –Опубл. 13.04.83. –РЖ Химия, 1984, 12Н29.
282. Авторское свидетельство СССР 740741. Способ получения гексафторацетона. / Я.М. Виленчик, Г.И. Якурнова, Г.И. Лекомцева, Л.П. Заякина, А.П. Харченко. –Опубл. 15.06.80. –РЖ Химия, 1980, 24Н67.
283. Авторское свидетельство СССР 859348. Способ получения гексафторацетона. / Я.М. Виленчик, Г.И. Якурнова. –Опубл. в Б.И., 1981, № 32, РЖ Химия, 1982, 21Н12.
284. Патент США 4238416. Способ изомеризации фторированных соединений. / Д. Кодуо. –Опубл. 09.12.80. –РЖ Химия, 1981, 17Н65.
285. Киперман С.Л. Введение в кинетику гетерогенных каталитических реакций. М.: Наука, 1964, с. 401.
286. Тюльга Г.М., Губанов В.А., Попова И.С., Петрухно Л.А., Болховец Б.М., Тройчанская П.Е., Долгопольский И.М. Изучение реакции фторангидридов перфторкарбоновых кислот с фторидами щелочных металлов. –ЖОрХ, 1973, т. 14, вып. 11, с. 2339-2346.
287. Коншин А.И., Губанов В.А. Об оценке эффективности способа очистки хладоновых растворов перфторполиоксаметиленацетилфторидов. –Отчет предприятия п/я В-8415.
288. Авторское свидетельство СССР 188404. Фторсополимер винилиденфторида с 3,6,8,10,12-пентаоксаперфтортридеценом для термоморозоагрессивостойких изделий. / Грейс А.М., Ершов А.Е., Захаров В.Ю. и др.
289. Карцов С.В., Валов П.И., Соколов Л.Ф., Соколов С.В. Роль поверхности реакционного сосуда в процессе жидкофазного окисления гексафторпропилена. –Изв. АН СССР, сер. хим., 1978, № 10. с. 2268-2272.
290. Авторское свидетельство 229185. Способ получения перфторполиоксаметиленацетилфторидов. / Захаров В.Ю., Гольдинов А.Л., Боровнев Л.М. и др.
291. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Динамика изменения состава газовой и жидкой фаз в промышленном реакторе синтеза окиси гексафторпропилена в ходе операции окисления. –Отчет предприятия п/я А-1619, 1983, инв. № 1089, 10с.
292. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Динамика образования перфторполиоксаметилен ацетилфторидов в процессе жидкофазного окисления гексафторпропилена кислородом на промышленной установке. –Отчет предприятия п/я А-1619, 1985, инв. № 1165, 22с.
293. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Окисление гексафторпропилена молекулярным кислородом в присутствии 1,2-дибромтетрафторэтана (хладона-114В2). Получение окиси гексафторпропилена. –Отчет предприятия п/я А-1619, 1982, инв. № 1072, 57с.
294. Авторское свидетельство СССР 1128436. Пламягасящая смесь. / Боровнев Л.М., Гусенков М.В., Жукова В.А. и др.
295. Кравченко Н.Н., Попов В.А., Ершов Ю.А., Померанцева Э.Г. Некоторые особенности фотолиза перфторированных полиэфиров. –ВМС, т. 23, № 3, 1981, с. 180-183.
296. Заявка СССР 3011822. Способ регенерации хладона-113. / Паниткова Е.С., Беренблит В.В., Сенюшов А.Н. и др.
297. Зейфман Ю.В., Тер-Габриэлян Е.Г., Ганбарян Н.П., Кнунянц И.Л. Химия перфторизобутилена. –Успехи химии, 1984, т. 53, вып. 3, с. 431-461.
298. Кнунянц И.Л., Шокина В.В., Кулешова А.Д. Присоединение галоидводородов к фторолефинам. –Изв. АН СССР, сер. хим., 1960, № 9, с.1693-1695.
299. Постовой С.А., Мысов Е.И., Зейфман Ю.В., Кнунянц И.Л. Нуклеофильная конденсация октафторизобутилена с тетрафторэтиленом. –Изв. АН СССР, сер. хим., 1982, № 7, с. 1586-1590.
300. Патент США 3389187. Димер перфторизобутилена. / Д.Л. Миллер. –Опубл. 18.06.68. –РЖ Химия, 1969, 17Н3211.
301. Кнунянц И.Л., Герман Л.С., Дяткин Б.П. Реакции фторолефинов. Сообщение ІІ. Взаимодействие соединений ряда перфторизобутилена с аминами и аммиаком. –Изв. АН СССР, отд.хим.н., 1960, № 2, с. 221-230.
302. Патент Японии 53-73504. Удаление октафторизобутилена. / Курода Такэси, Фурукава Йосики, Мацуоха Ну. –Опубл. 30.06.78. РЖ Химия, 1979, 10Н2011.
303. Дорогимский В.А., Коломиец А.Ф., Сокольский Г.А. О реакции октафторизобутилена с этиленгликолем и глицерином. –ЖОрХ, 1983, т.19, вып. 8, с.1762-1763.
304. Авторское свидетельство СССР 129653. Способ получения гексафторизомасляной кислоты. / Кнунянц И.Л., Чебурков Ю.А., Красуская М.П. –РЖ Химия, 1961, 9Л75.
305. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Промышленные испытания и внедрение способа очистки газов пиролиза тетрафторэтилена от перфторизобутилена. –Отчет предприятия п/я А-1619, 1981, инв. № 1048, 34с.
306. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Увеличение степени использования сырья в производстве гексафторпропилена путем более полного извлечения октафторциклобутана из кубовой фракции. –Отчет предприятия п/я А-1619, 1982, инв. № 1063, 16с.
307. Авторское свидетельство СССР 180854. Способ получения гексафторпропилена. / Боровнев Л.М., Виноградова А.П., Голубев А.Н. и др.
308. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Комплексная переработка кубовой фракции производства тетрафторэтилена. –Отчет предприятия п/я А-1619, 1985, инв. № 15/107 ДСП, 17с.
309. Авторское свидетельство СССР 1336492. Способ получения 2-хлортетрафторэтилфторсульфата. / Фокин А.В., Рапкин А.И., Татаринов А.С. и др.
310. Авторское свидетельство СССР 1184808. Способ получения монофторсульфата брома. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
311. Авторское свидетельство СССР 1239092. Способ получения трис(фторсульфата) брома. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
312. Заявка СССР 3982041. 3-(Полифторпропионамидо) пропил-2-оксиэтилдиметиламмоний хлориды, обладающие гипотензивным действием. / Фокин А.В., Ковалев Г.В., Мазанов Л.С. и др.
313. Заявка СССР 3982040. N-Полифторацильные производные γ-амномасляной кислоты, обладающие гипертензивным действием. / Фокин А.В., Ковалев Г.В., Мухина Н.В. и др. Пол. реш. от 26.09.86.
314. Автотское свидетельство СССР 1334655. 3-(ω-Хлорперфторалканоиламидо)пропил-диметил-2-оксиэтиламммоний хлориды в качестве поверхностно-активных веществ. /Фокин А.В., Рапкин А.И., Студнев Ю.Н. и др.
315. Авторское свидетельство СССР 196359. Способ получения ω-хлорперфторкарбоновых кислот и эмульгатор. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
316. Патент Германии 3034549. Способ получения перфторированных фторангидридов карбоновых кислот. / Вернер Ш. –Опубл. 29.04.82, –РЖ Химия, 1983, 17Н5711.
317. Промышленные катализаторы и носители. Справочник. –Новосибирск, 1972, с. 246.
318. Т. Ван дер Плас. Текстура и химия поверхности углеродных тел. В кн.: Строение и свойства адсорбентов и катализаторов. –М.: Мир, 1973, с. 436-441.
319. Патрик С. Успехи химии фтора. –М., Л., Химия, 1964, т.1, № 2, с. 336-379.
Материал рекомендован к публикации членом редколлегии С.М. Игумновым
Fluorine Notes, 2014, 96, 1-2
