Received: март 2014
Fluorine Notes, 2014, 96, 5-6
Структурные свойства, Теория функционала плотности (DFT), расчёт зарядов NBO для C18H10F11BrN4O
Shima Rahmani*,1, Shahriar Ghammami2, Amir Lashgari2
1Department of Chemistry, Faculty of Science, Azad University unit Of Takestan, Takestan, Iran.
2Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin, Iran.
*Corresponding Author E-mail: Ikiu2014@yahoo.com
Аннотация. Результаты вычислений B3LYP/LANL2DZ показали некоторые величины длины связей и углов для C18H10F11BrN4O. в данной статье оптимизированные геометрии и частоты стационарных точек нового вещества с химической формулой C18H10F11BrN4O вычислены с помощью методов DFT с базовым набором 6-311 и LANL2DZ.. ЯМР спектры C18H10F11BrN4O вычислялись с помощью методов DFT (B3LYP) с базовыми наборами LANL2DZ.
Ключевые слова: Электронная структура, фторное соединение, вычисления DFT , ЯМР, вибрационный анализ.
Введение
Fluorous chemistry - это относительно новый термин, который подразумевает использование перфторированных соединений или перфторированных растворителей для того, чтобы упростить извлечение катализаторов или реакционных продуктов. Наличие перфторированной группы придает соединению уникальные физические свойства, в первую очередь способность растворяться в перфторорганических растворителях.
Молекулы таких соединений имеют органический фрагмент и сильно фторированный фрагмент. В идеале, органический фрагмент управляет реакционной способностью а фторированный фрагмент позволяет контролировать выделение таких соединений. Данныее свойства моутт быть полезными для органического синтеза и методов разделения, таких как экстракция твердой фазы. [1] Слово «Fluorous» относительное новое, но уже завоевало общее признание. В качестве прилагательного «Fluorous» относят к сильно фторированным насыщенным органическим материалам, молекулам или молекулярным фрагментам. Историю появления и развития нового термина см. [2].
Такие технологии (fluorous technologies) повзоляют проводить процессы с высокой скоростью. [3-5] .
Одно из таких соединений рассмотрено в настоящей статье.
Структура соединения оптимизирована с использованием метода DFT (B3LYP) с базисными наборами LANL2DZ с помощью программы Gaussian 09w [6]. Методы теории функциональной плотности использовались для определения оптимизированных структур C18H10F11BrN4O. Первоначальные вычисления выполнялись на уровне DFT и использовались базисные наборы LANL2DZ. Локальные минимумы были получены с помощью полной геометрической оптимизации [7].
РАСЧЕТЫ
Оптимизированные структурные параметры использовались при вычислении вибрационной частоты на уровне DFT, чтобы характеризировать все стационарные точки как минимальные. Все компьютерные вычисления проводились с использованием программы Gaussian 09w. Гармонические вибрационные частоты (ν) в cm-1 и инфракрасные интенсивности (int) в километрах на моль всех соединений выполнялись на том же уровне на соответствующих полностью оптимизированных измерениях. Молекулярные геометрии минимума энергии были обнаружены минимизацией энергии в соответствие со всеми геометрическими координатами без налагания любых симметрических ограничений.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Молекулярные Свойства
Структуры соединений показаны на Рис.1. Все вычисления выполнены с помощью компьютерной программы Gaussian 09w.
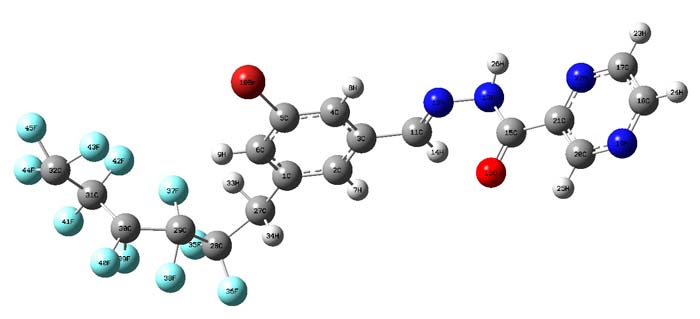
Рис 1. TСтруктура молекулы C18H10F11BrN4O.
Теоретические вычисления связей и углов соединения определялись оптимизацией геометрии (измерений) (Таблица 1). Вычисленные гибридизации NBO показаны в Таблице 2. Результаты вычислений B3LYP/ LANL2DZ показали, что величины длины связи (C5-Br10) для C18H10F11BrN4O - 1.90 Å соответственно и величины длины связи (N12-N13) для C18H10F11BrN4O - 1.39 Å соответственно.
Вычисленные гибридизации NBO - это существенные параметры нашего исследования. Структура соединения была оптимизирована с помощью метода DFT (B3LYP) с базовыми наборами LANL2DZ с использованием программы Gaussian 09w. Методы теории функциональной плотности применялись для определения оптимизированных структур C18H10F11BrN4O (Таблица 1, Рисунок 1). Эти естественные локализованные наборы являются промежуточными звеньями между базисными атомными орбиталями (AO) и молекулярными орбиталями (MO): атомная орбиталь→ NAO→ NHO→ NBO→ NLMO→ Молекулярная орбиталь, Естественная (локализованная) орбиталь использовались в вычислительной химии для вычисления распределения электронной плотности в атомах и в связях между атомами.
Table 1. Геометрические параметры C18H10F11BrN4O некоторых длин связей (Å)и углов (°).
|
Угол (°) |
Связь |
Длина (Å) |
Связь |
|
120.04 |
Br10-C5-C4 |
1.90 |
C5-Br10 |
|
120.30 |
C11-N12-N13 |
1.29 |
C11-N12 |
|
120.04 |
N14-C15-C21 |
1.39 |
N12-N13 |
|
119.94 |
H23-C17-C18 |
1.25 |
C15-O16 |
|
119.95 |
C20-N19-C18 |
1.33 |
C21-N22 |
|
109.32 |
F37-C29-C30 |
1.34 |
C32-F43 |
|
140.49 |
F43-C32-F45 |
1.34 |
C28-F35 |
NBO Исследование (Орбиталь естественной связи) структур
Вычисленные гибридизации NBO - это существенные параметры нашего исследования. Структура соединения была оптимизирована с помощью метода DFT (B3LYP) с базовыми наборами LANL2DZ с использованием программы Gaussian 09w. Методы теории функциональной плотности применялись для определения оптимизированных структур C18H10F11BrN4O (Таблица 1, Рисунок 1). Эти естественные локализованные наборы являются промежуточными звеньями между базисными атомными орбиталями (AO) и молекулярными орбиталями (MO): атомная орбиталь→ NAO→ NHO→ NBO→ NLMO→ Молекулярная орбиталь, Естественная (локализованная) орбиталь использовались в вычислительной химии для вычисления распределения электронной плотности в атомах и в связях между атомами.
С помощью программы способной вычислять NBOs, можно найти оптимальные структуры связи [8]. Оптимальная структура Льюиса может определяться как структура с максимальным количеством электронного заряда на орбиталях Льюиса (Заряд Льюиса) [9].
Низкий электронный заряд на орбиталях Льюиса указывает на наличие сильного влияния электронной делокализации. В структурах резонанса могут существовать главные и второстепенные структуры. Например, для амидов вычисления NBO показывают, что структура с двойной связью карбонила доминирует в структуре Льюиса. Однако, в вычислениях NBO «ковалентно-ионного резонанса» не требуется включать эффекты полярности связей в структуры резонанса [10]. Это идентично другим современным методам теории валентных связей.
Орбитали естественной связи (NBOs) – это локализованные орбитали с несколькими центрами, которые описывают подобные Льюисовским схемы поведения молекулярных связей пар электронов в оптимально компактной форме. Более точно орбитали естественной связи NBOs – это орто-нормальный набор локализованных орбиталей «максимальной занятости», чьи ведущие N/2 члены (или N члены в случае незамкнутой оболочки) дают наиболее точное возможное описание типа Льюисовского полной плотности N-электрона. Этот анализ выполнен с помощью проверки всех возможных взаимодействий между «заполненными» (донорными) Льюисовского типа NBOs и «пустыми» (акцепторными) не-льюисовскими NBOs, и с помощь оценки их энергетической значимости теорией пертуберации второго порядка. Так как эти взаимодействия приводят к передаче занятости от локализованных NBOs идеализированной структуры Льюиса к пустым нельюисовким орбиталям (и таким образом, к отправлениям от описания идеализированной структуры Льюиса), они называются коррекцией «делокализации» естественной структуры Льюиса нулевого порядка.
Естественные заряды были вычислены с помощью модуля естественной орбитали, вставленного в программу Gaussian 09w. Данные количества получены из анализа популяции (занятости) NBO. Последний обеспечивает картину орбитали, которая ближе к классической структуре Льюиса. Анализ NBO затрагивающий гибридизации избранных связей вычисляли методом B3LYP с базовым набором LANL2DZ y (Табл. 2).
Таблица 2. NBO рассчитанные гибридизации для C18H10F11BrN4O (B3LYP/LANL2DZ).
|
Связь |
Атом |
B3LYP |
|
C-C |
C1-C2 |
S1P1.81 S1P1.94 |
|
C-F |
C28-F36 |
S1P3.22,S1P2.67 |
|
C-H |
C2-H7 |
S1P0,S1P2.50 |
|
C-Br |
C5-Br10 |
S1P3.71,S1P6.34 |
|
C-N |
C17-N22 |
S1P2.25,S1P1.83 |
Эти данные показывают, что существует гиперсопряжение электронов между лиганд атомами и центровым атомом металла. Данные сопряжения основываются на p-d π-связях. Вычисляемая гибридизация NBO для C18H10F11BrN4O демонстрирует, что все соединения обладают SPX гибридизацией и непланарными конфигурациями. Тотальная гибридизация этих молекул – это SPX , что подтверждается структурно. Число гибридизаций связей демонстрирует неравенство между углами центровых атомов (Табл. 2) показывающими искажение нормальных VSEPR структур и подтверждающими девиацию от VSEPR структур. (Рис. 1).
ИК спектр и спектр комбинационного рассеивания (Рамана)
В данной работе мы использовали компьютерную программу Gaussian 09w, способную получить инфракрасный спектр (Рисунок 2 и Таблица 3). Вычисление колебательных частот тесно связано с процедурой структурной оптимизации в практике исследования. Они используются для стимулирования инфракрасного и спектра комбинационного рассеивания. Спектр комбинационного рассеивания показан на Рисунке 3. Спектроскопия комбинационного рассеивания – это техника спектроскопии, используемая для наблюдения вибрационных, вращательных и других низкочастотных моделей в системе [13]. Эффект комбинационного рассеивания света случается, когда свет ударяется о молекулу и взаимодействует с облаком электрона и связями этой молекулы.
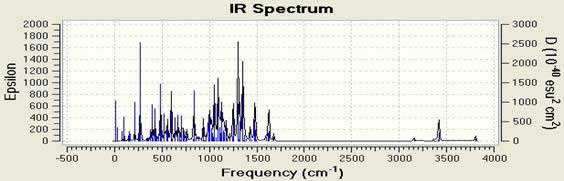
Fig 2.Раасчитанный ИК спектр C18H10F11BrN4O .
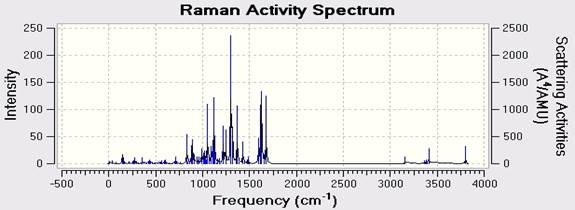
Fig. 3. Раасчитанный Рамановский спектр C18H10F11BrN4O .
Таблица 3. DFT (B3LYP) with LANL2DZ level calculated vibrational frequencies of C18H10F11BrN4O (cm -1).
|
Ипсилон |
Частота |
ИК |
Спектр Рамана |
|
1700 |
273.49 |
173.49 |
108.75 |
|
700 |
6.41 |
1.67 |
44.66 |
|
400 |
96.71 |
14.79 |
6.45 |
|
900 |
1053.63 |
348.54 |
1109.55 |
Спектры ЯМР
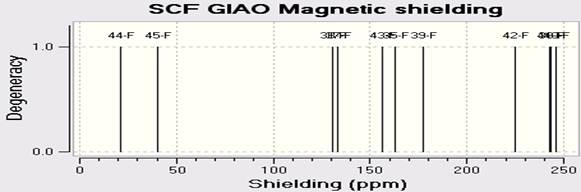
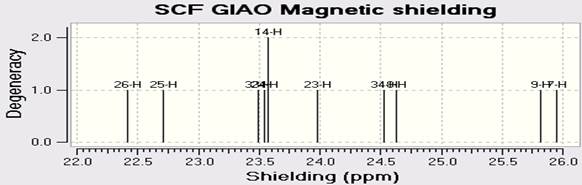
ЯМР спектры C18H10F11BrN4O были рассчитаны методом DFT(B3LYP) с применением базового набора LANL2DZ. Спектры C13-ЯМР, H-ЯМР (ПМР) и F19-NMR представлены на Рисунках 4a-c.
 a)
a)
 b)
b)
 c)
c)
Fig. 4. Рассчитанные A) C-ЯМР B) F-ЯМР C) H-ЯМР спектры C18H10F11BrN4O Z.
Граничная молекулярная орбиталь
Мы концентрируем внимание на этих двух орбиталях, так как они наиболее близки по энергии. Данные орбитали участвуют в химической реактивности на тесном уровне, так как они наиболее доступны для электрофилов и нуклеофилов соответственно. Другое главное изменение связано с граничными орбиталями, (HOMO) и *(LUMO) орбиталями [11-12].
HOMO (ВЗМО, высокоэнергетическая заселённая молекулярная орбиталь) представляет способность отдавать электрон, LUMO (низшая незанятая молекулярная орбиталь) в качестве акцептора электронов представляет способность получать электрон. Энергии HOMO и LUMO вычислялись с помощью метода B3LYP/LANL2DZ. Данная абсорбция электронов соответствует переходу от начального к первому возбужденному состоянию и, главным образом, описывается возбуждением одного электрона с высшей занятой молекулы или орбитали (LUMO), обе, т.е. высокоэнергетическая заселённая молекулярная орбиталь (HOMO) и низшая незанятая молекулярная орбиталь (LUMO) – это главные орбитали, принимающие участие в химической стабильности. Поэтому, в то время как энергия HOMO напрямую относится к потенциалу ионизации, энергия LUMO напрямую относится к электронному сродству. Энергетическая разница между орбиталями HOMO и LUMO называется энергетическим зазором, что является важным для стабильности структур. Дополнительно 3D схемы высших занятых молекулярных орбиталей (HOMOs) и низших незанятых молекулярных орбиталей (LUMOs) показаны на Рисунке 5.
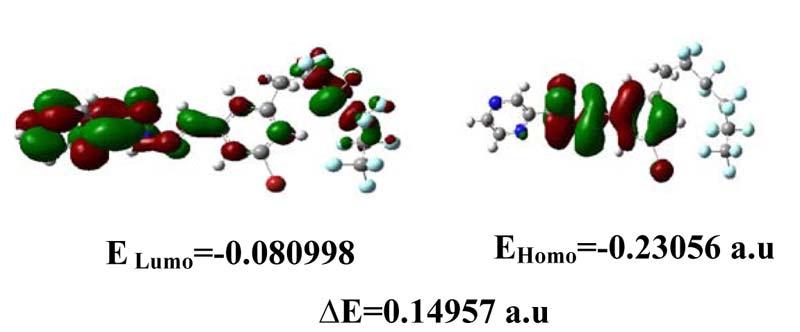
Рис 5. Граничные молекулярные орбитали для C18H10F11BrN4O по теории LANL2DZ.
Распределение плотности электронов
Распределение полной плотности электронов – это физическое свойство молекул. Плотность электронов обычно показывается как сравнение определенной электронной плотности с предсказанной с помощью сферических моделей атомов, и она называется деформационной электронной плотностью. Полная электронная плотность вычислена с помощью теории функциональной плотности DFT (B3LYP)/ LANL2DZ. Контурные карты электронной плотности для C18H10F11BrN4O изображены на рисунке 6. Также дополнительно (Таблица 4) даны заряды атомов C18H10F11BrN4O, а структура атомных зарядов дана на рисунке 7.
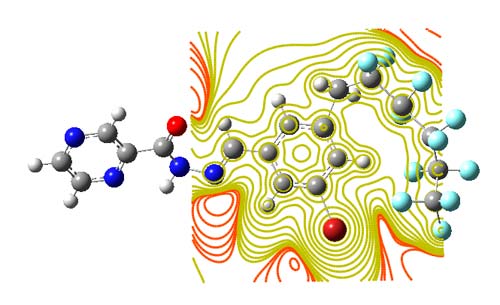
Рис. 6. Карты электронной плотности для C18H10F11BrN4O рассчитанные с помощью DFT/LANL2DZ .
Рис. 7. Структура зарядов атомов C18H10F11BrN4O .
Таблица 4. Зарядв атомов в C18H10F11BrN4O.
|
Atom |
Заряд |
|
C5 |
-2.292 |
|
Br10 |
0.918 |
|
N12 |
-0.079 |
|
N1 |
-0.302 |
|
O16 |
0.255 |
|
F37 |
-0.212 |
|
F42 |
-2.102 |
|
H14 |
0.254 |
ЗАКЛЮЧЕНИЕ
В данной статье оптимизированные геометрии и частоты стационарной точки и пути минимальной энергии вычислены с помощью методов теории функциональной плотности DFT (B3LYP) с базисными наборами LANL2DZ. В данной статье инфракрасный спектр, спектр комбинационного рассеивания света и ЯМР спектры C18H10F11BrN4O получены с применением теоретических методов [14-17].
БЛАГОДАРНОСТЬ
Работа выполнена при финансовой поддержке Research Council of Imam Khomeini International University.
Список литературы
- Gladysz, J. A.; Curran, D. P.; Horvath, I. T. Eds. f Fluorous Chemistry Handbook Wiley, Wiley--VCH, 2004VCH, 2004.
- Gladysz, J. & Curran, D., “Fluorous chemistry: from biphasic catalysis to a parallel universe and beyond”, Tetrahedron, 58, (2002), 3823-3825.
- W.Zhang, Chem. Rev. 2004, 104, 2531.104, 2531.
- Ellman, J., et al.J. Org. Chem. 1997, 62, 1242J. 1242
- Zhang, W. “Fluorous Protecting Groups and Tags” in Handbook of Fluorous Chemistry Gladysz, J. A.; Curran, D. P.; Horvath, I. T. Eds. Wiley-VCH, 2004, pp222-2362.
- Frisch, M. J. Trucks, G. W. 1998. GASSIAN 98 (Revision A. 3) Gaussian Inc., Pittsburgh, PA, USA.
- Ghammamy, Sh., K. Mehrani, Rostamzadehmansor, S. and Sahebalzamani, H. 2011. Density functional theory studies on the structure, vibrational spectra of three new tetrahalogenoferrate (III) complexes. Natural Science, 3, 683-688.
- Weinhold, Frank. and R. Landis, Clark, 1964. Discovering Chemistry with Natural Bond Orbitals. New Jersey: John Wiley & Sons., Pp: 132-133. ISBN 978-1-118-22916-3.
- Frank Weinhold and R. Clark, 2001. Natural bond Orbital and extensions of localized bonding concepts Chem Educ. Res. Pract. Eur., 2: 91-104.
- Gottimukkala Rambabu, Zeenat Fatima, Bandapalli Palakshi Reddy, Vijayapar Tthasarathi and Vijayakumar, 2000. Stereoelectronic Interactions in Cyclohexane, 1, 3-Dioxane,1, 3-Oxathiane and 1,3-Dithiane: W-Effect, C-XTó C-H Interactions, Anomeric Effects What Is Really Important? J. Org. Chem., 65: 3910-3919.
- Seidy and Shahriar Ghammamy, 2012. Structural Properties, Natural Bond Orbital, Theory Functional Calculations (DFT) and Energies for the Halorganic Compounds Current World Environment, 7(2): 221-226.
- Jursic, B.S., 2000. A B3LYP hybrid density functional theory study of structural properties,and energies heats of formation for silicon–hydroge compounds, Journal of Molecular Structure (Theochem), 497: 65-73.
- Gardiner, D.J., 1989. Practical Raman spectroscopy. Springer-Verlag. ISBN 978-0- 387-50254-0.
- Abou-Deif, M.H., M.A. Rashed, M.A.A. Sallam, E.A.H. Mostafa and W.A. Ramadan, 2013, Characterization of Twenty Wheat Varieties by ISSR Markers, Middle-East Journal of Scientific Research, 15(2): 168-175.
- Kabiru Jinjiri Ringim, 2013. Understanding of Account Holder in Conventional Bank toward Islamic Banking Products, Middle-East Journal of Scientific Research, 15(2): 176-183.
- Muhammad Azam, Sallahuddin Hassan and Khairuzzaman, 2013. Corruption, Workers Remittances, Fdi and Economic Growth in Five South and South East Asian Countries: A Panel Data Approach Middle-East Journal of Scientific Research, 15(2): 184-190.
- Lewis, D. F. V., C. Ioannides, and Parke, D. V. 1994. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: computer analysis of structure- activity relationships. Xenobiotica, 24: 401-408.
Статья рекомендована к публикации членом редколлегии д.х.н. С.М. Игумновым
Fluorine Notes, 2014, 96, 5-6
