Received: March , 2014
Fluorine Notes, 2014, 96, 5-6
Structural Properties, Density Functional Theory (DFT), Natural Bond Orbital and Energy Calculations for the Fluorous compound: C18H10F11BrN4O
Shima Rahmani*,1, Shahriar Ghammami2, Amir Lashgari2
1Department of Chemistry, Faculty of Science, Azad University unit Of Takestan, Takestan, Iran.
2Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin, Iran.
*Corresponding Author E-mail: Ikiu2014@yahoo.com
Abstract: B3LYP/LANL2DZ calculation results indicated some selected bond length and bond angles values for the C18H10F11BrN4O . In this article, the optimized geometries and frequencies of the stationary point and the minimum-energy paths of new compound with C18H10F11BrN4O chemical formula are calculated by using the DFT methods with 6-311 and LANL2DZ basis set. The NMR spectra of C18H10F11BrN4O is calculated by using the DFT (B3LYP) methods with LANL2DZ basis sets.
Key words: Electronic structure, Fluorous compound, DFT Calculations, NMR, Vibrational analysis
Introduction
Fluorous molecules comprise an organic domain (chain) and a highly fluorinated domain (chain). Ideally, the organic domain controls reactivity and the fluorinated domain controls separation. The aim is to facilitate the separation process. Fluorous chemistry or Fluorocarbon and Organofluorine chemistry involves the use of perfluorinated compounds or perfluorinated substituents to facilitate recovery of a catalyst or reaction product. This property can be useful in organic synthesis and separation methods such as solid phase extraction [1]. The word "fluorous" is relatively new but has gained general acceptance. As an adjective, “fluorous” refers to highly fluorinated saturated organic materials, molecules, or molecular fragments. The history of "fluorous chemistry" definition one can see in [2].
Integrated fluorous technologies with microwave reactions and multi component reactions provides valuable tools for Fluorous high-throughput organic synthesis. [3-5]
The structure of the compound has been optimized by using the DFT (B3LYP) method with the LANL2DZ basis sets, using the Gaussian 09w [6] program. Density functional theory methods were employed to determine the optimized structures of C18H10F11BrN4O. Initial calculations were performed at the DFT level and split — valence plus polarization LANL2DZ basis sets were used. Local minima were obtained by full geometrical optimization having all positive frequencies. [7]
COMPUTATIONAL
The optimized structural parameters were used in the vibrational frequency calculations at the DFT level to characterize all stationary points as minima. All computational are carried out using Gaussian 09w program. Harmonic vibrational frequencies (ν) in cm-1 and infrared intensities (int) in Kilometer per mole of all compounds were performed at the same level on the respective fully optimized geometries. Energy minimum molecular geometries were located by minimizing energy, with Respect to all geometrical coordinates without imposing any symmetrical constraints.
RESULTS AND DISCUSSION
Molecular properties
The structure of compound is shown on Figure 1. All calculations were carried out using the computer program Gaussian 09w.
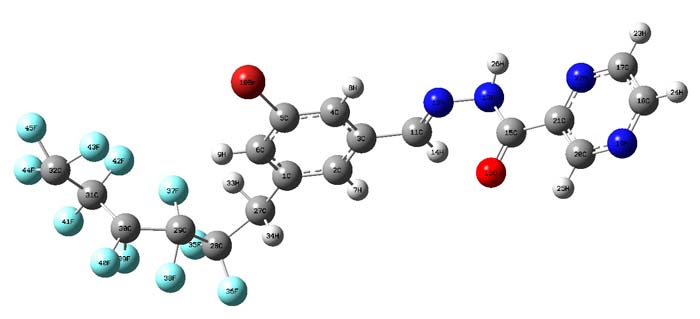
Fig 1. The schematic structure of the C18H10F11BrN4O.
Theoretical calculation both bonds and angles of compound were determined by optimizing the geometry (Table 1). The NBO Calculated Hybridizations are reported in Table 2. B3LYP/ LANL2DZ calculation results showed that the (C5-Br10) bond length values for the C18H10F11BrN4O are 1.90 Е respectively. And (N12-N13) bond length values for the C18H10F11BrN4O are 1.39 Е respectively.
NBO (Natural Bond Orbital) study on structures
NBO Calculated Hybridizations are significant parameters for our studying. The structure of the compound has been optimized by using the DFT (B3LYP) method with the LANL2DZ basis sets, using the Gaussian 09w program. Density functional theory methods were employed to determine the optimized structures C18H10F11BrN4O (Table 1, Figure 1). These natural localized sets are intermediate between basis atomic orbitals (AO) and molecular orbitals (MO): Atomic orbital → NAO→ NHO→ NBO→ NLMO→ Molecular orbital. Natural (localized) orbitals are used in computational chemistry to calculate the distribution of electron density in atoms and bonds between atoms.
Table 1. Geometrical parameters optimized for C18H10F11BrN4O some selected bond lengths (Е) and angles (°).
|
angles (°) |
Bond |
lengths (Е) |
Bond |
|
120.04 |
Br10-C5-C4 |
1.90 |
C5-Br10 |
|
120.30 |
C11-N12-N13 |
1.29 |
C11-N12 |
|
120.04 |
N14-C15-C21 |
1.39 |
N12-N13 |
|
119.94 |
H23-C17-C18 |
1.25 |
C15-O16 |
|
119.95 |
C20-N19-C18 |
1.33 |
C21-N22 |
|
109.32 |
F37-C29-C30 |
1.34 |
C32-F43 |
|
140.49 |
F43-C32-F45 |
1.34 |
C28-F35 |
They have the "maximum-occupancy character" in localized 1-center and 2-center regions of the molecule [8]. With a computer program that can calculate NBOs and optimal Lewis structures can be found. An optimal Lewis structure can be defined as that one with the maximum amount of electronic charge in Lewis orbitals (Lewis charge) [9].
A low amount of electronic charge in Lewis orbitals indicates strong effects of electron delocalization. In resonance structures, major and minor contributing structures may exist. For amides, for example, NBO calculations show that the structure with a carbonyl double bond is the dominant Lewis structure. However, in NBO calculations, "covalent-ionic resonance" is not needed due to the inclusion of bond-polarity effects in the resonance structures [10]. This is similar to other modern valence bond theory methods.
Natural Bond Orbital's (NBOs) are localized few-center orbital's that describe the Lewis-like molecular bonding pattern of electron pairs in optimally compact form. More precisely, NBOs are an ortho-normal set of localized "maximum occupancy" orbital's whose leading N/2 members (or N members in the open-shell case) give the most accurate possible Lewis-like description of the total N-electron density. This analysis is carried out by examining all possible interactions between "filled" (donor) Lewis-type NBOs and "empty" (acceptor) non-Lewis NBOs, and estimating their energetic importance by 2nd-order perturbation theory. Since these interactions lead to donation of occupancy from the localized NBOs of the idealized Lewis structure into the empty non-Lewis orbitals (and thus, to departures from the idealized Lewis structure description), they are referred to as "delocalization" corrections to the zeroth-order natural Lewis structure.
Natural charges have been computed using natural bond orbital (NBO) module implemented in Gaussian 09w. These quantities are derived from the NBO population analysis. The former provides an orbital picture that is closer to the classical Lewis structure. The NBO analysis involving hybridizations of selected bonds are calculated at B3LYP methods LANL2DZ level of theory (Table 2).
Table 2. The NBO Calculated Hybridizations for C18H10F11BrN4O (B3LYP/LANL2DZ)
|
Bond |
Atom |
B3LYP |
|
C-C |
C1-C2 |
S1P1.81 S1P1.94 |
|
C-F |
C28-F36 |
S1P3.22,S1P2.67 |
|
C-H |
C2-H7 |
S1P0,S1P2.50 |
|
C-Br |
C5-Br10 |
S1P3.71,S1P6.34 |
|
C-N |
C17-N22 |
S1P2.25,S1P1.83 |
These data shows the hyper conjugation of electrons between ligand atoms with central metal atom. These conjugations stand on the base of p-d π-bonding. The NBO calculated hybridization for C18H10F11BrN4O shows that all of compounds have SPX hybridization and non planar configurations. The total hybridization of these molecules are SPX that confirmed by structure. The amount of bond hybridization showed is equality between central atoms angles (Table 2) shown distortion from normal VSEPR structures and confirmed deviation from VSEPR structures. (Figure 1).
IR and Raman spectra
Infrared spectral range of 12800-10 cm-1 spectral region is extensive. The infrared spectrum a molecule to be absorbed by the dipole moment of the molecule should be expected to change as a result of vibration and rotation. Infrared spectrum gives information about the structure of a molecule. The spectrum of basic information about the various functional groups as well as any other information that may be used to identify substances implies.
In this article we use the Gaussian 09w software able to obtain infrared spectrum (Figure 2 & Table 3). Closely connected in research practice to the procedure of structural optimization is the calculation of vibrational frequencies. They are used for simulating infrared (IR) or Raman spectra. You can see in Figure 3 Raman spectra. Raman spectroscopy is a spectroscopic technique used to observe vibrational, rotational and other low-frequency modes in a system [13]. The Raman Effect occurs when light impinges upon a molecule and interacts with the electron cloud and the bonds of that molecule. Generally under red and Raman spectrum are complementary. Because each of them with a different set of vibrational states of a molecule are associated with. A major advantage compared to Raman Spectroscopy Infrared spectrum that does not overlap the water. Therefore in the Raman spectra of aqueous solutions can be achieved.
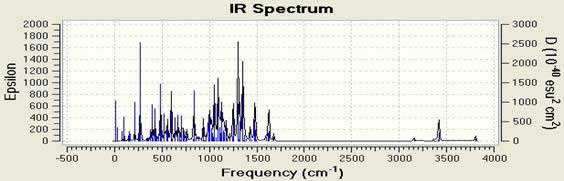
Fig 2. Calculated infrared spectra of C18H10F11BrN4O − (top to bottom, Frequencies in cm−1, intensities in arbitrary units).
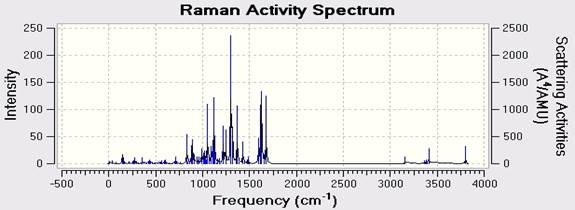
Fig. 3. Calculated Raman spectra of C18H10F11BrN4O by DFT (B3LY)/LANL2DZ.
Table 3. DFT (B3LYP) with LANL2DZ level calculated vibrational frequencies of C18H10F11BrN4O (cm -1).
|
Epsilon |
Freq |
Infrared |
Raman Activity |
|
1700 |
273.49 |
173.49 |
108.75 |
|
700 |
6.41 |
1.67 |
44.66 |
|
400 |
96.71 |
14.79 |
6.45 |
|
900 |
1053.63 |
348.54 |
1109.55 |
NMR spectra
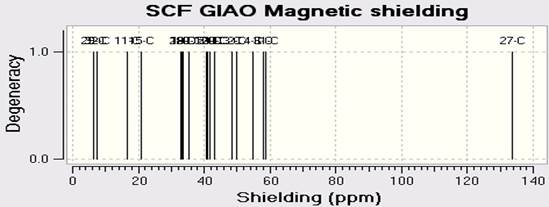
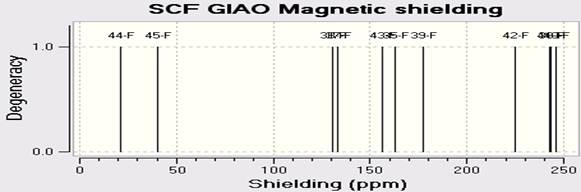
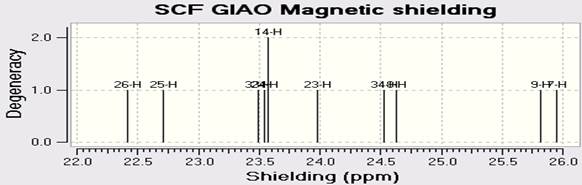
NMR spectroscopy is used for the atoms of the nucleus relative to the magnetic field sensitive. NMR spectroscopy is a useful technique for identifying and analyzing organic compounds. In this we have carried out computed chemical shift calculations of present molecule. Whereas infrared spectroscopy provides information on the functional groups present in the molecule goes nuclear magnetic resonance will inform us of the number of each type of hydrogen. In this article, NMR Spectra of C18H10F11BrN4O are calculated by using the DFT (B3LYP) methods with LANL2DZ basis sets. The spectra C-NMR, H-NMR, and F-NMR are in Figures 4a-c.
 a)
a)
 b)
b)
 c)
c)
Fig. 4. Calculated A) C-NMR B) F-NMR C) H-NMR spectra of C18H10F11BrN4O by DFT (B3LY)/LANL2DZ.
Frontier molecular orbital
We concentrate attention on these two orbitals because they are closest in energy. These orbitals are intimately involved in chemical reactivity, because they are the most available to electrophiles and nucleophiles, respectively. Another key change has to do with the frontier orbitals, the (HOMO) and *(LUMO) orbitals [11-12].
The HOMO represents the ability to donate an electron, LUMO as an electron acceptor represents the ability to obtain an electron. The HOMO and LUMO energy were calculated by B3LYP/ LANL2DZ method. This electronic absorption corresponds to the transition from the ground to the first excited state and is mainly described by one electron excitation from the highest occupied molecular or orbital (LUMO) both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the main orbital take part in chemical stability. Therefore, while the energy of the HOMO is directly related to the ionization potential, LUMO energy is directly related to the electron affinity. Energy difference between HOMO and LUMO orbital is called as energy gap that is an important stability for structures. In addition, 3D plots of highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) are shown in Figure 5.
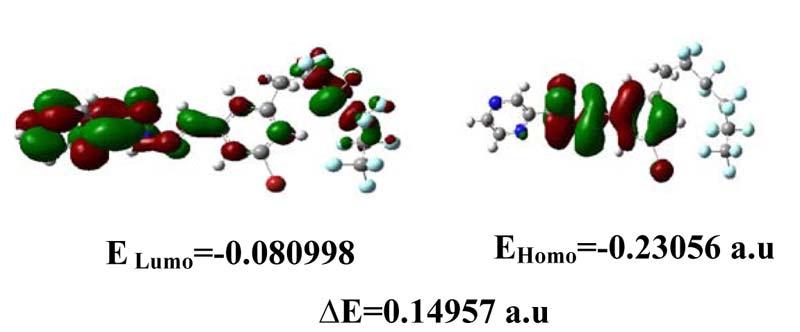
Fig 5. The atomic orbital of the frontier molecular orbital for C18H10F11BrN4O LANL2DZ level of theory.
Electron Density Distribution
The total electron density distribution is a physical property of molecules. The electron density is typically showed as a comparison of the identified electron density with that predictable by spherical models of the atoms and is called deformation electron density. The total electron density was calculated by DFT (B3LYP)/ LANL2DZ. The contour maps of electron density for C18H10F11BrN4O is shown in Figure 6. Also in addition (Table 4) given C18H10F11BrN4O atomic charges. The structure of atomic charges are shown in Figure 7.
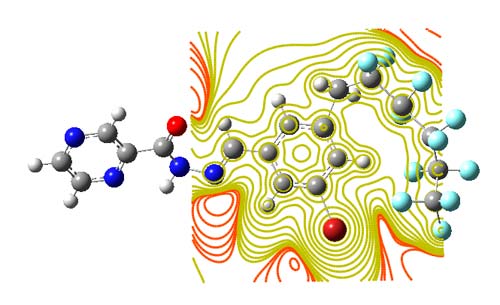
Fig 6. Contour maps of electron density for C18H10F11BrN4O was calculated at the DFT/LANL2DZ level.
Fig 7. The structure atomic charges of C18H10F11BrN4O with display charge distribution.
Table 4. Charges of C18H10F11BrN4O.
|
Atom NO |
Charge |
|
C5 |
-2.292 |
|
Br10 |
0.918 |
|
N12 |
-0.079 |
|
N1 |
-0.302 |
|
O16 |
0.255 |
|
F37 |
-0.212 |
|
F42 |
-2.102 |
|
H14 |
0.254 |
CONCLUSION
In this article the optimized geometries and frequencies of the stationary point and the minimum-energy paths are calculated by using the DFT (B3LYP) methods with LANL2DZ basis sets. This calculation results indicated some selected bond length and bond angles values for the C18H10F11BrN4O. The infrared spectra, Raman spectra and NMR of the C18H10F11BrN4O were studied using the theoretical methods [14-17].
ACKNOWLEDGMENTS
We gratefully acknowledge the financial support from the Research Council of Imam Khomeini International University.
References
- Gladysz, J. A.; Curran, D. P.; Horvath, I. T. Eds. f Fluorous Chemistry Handbook Wiley, Wiley--VCH, 2004VCH, 2004.
- Gladysz, J. & Curran, D., “Fluorous chemistry: from biphasic catalysis to a parallel universe and beyond”, Tetrahedron, 58, (2002), 3823-3825.
- W.Zhang, Chem. Rev. 2004, 104, 2531.104, 2531.
- Ellman, J., et al.J. Org. Chem. 1997, 62, 1242J. 1242
- Zhang, W. “Fluorous Protecting Groups and Tags” in Handbook of Fluorous Chemistry Gladysz, J. A.; Curran, D. P.; Horvath, I. T. Eds. Wiley-VCH, 2004, pp222-2362.
- Frisch, M. J. Trucks, G. W. 1998. GASSIAN 98 (Revision A. 3) Gaussian Inc., Pittsburgh, PA, USA.
- Ghammamy, Sh., K. Mehrani, Rostamzadehmansor, S. and Sahebalzamani, H. 2011. Density functional theory studies on the structure, vibrational spectra of three new tetrahalogenoferrate (III) complexes. Natural Science, 3, 683-688.
- Weinhold, Frank. and R. Landis, Clark, 1964. Discovering Chemistry with Natural Bond Orbitals. New Jersey: John Wiley & Sons., Pp: 132-133. ISBN 978-1-118-22916-3.
- Frank Weinhold and R. Clark, 2001. Natural bond Orbital and extensions of localized bonding concepts Chem Educ. Res. Pract. Eur., 2: 91-104.
- Gottimukkala Rambabu, Zeenat Fatima, Bandapalli Palakshi Reddy, Vijayapar Tthasarathi and Vijayakumar, 2000. Stereoelectronic Interactions in Cyclohexane, 1, 3-Dioxane,1, 3-Oxathiane and 1,3-Dithiane: W-Effect, C-XTу C-H Interactions, Anomeric Effects What Is Really Important? J. Org. Chem., 65: 3910-3919.
- Seidy and Shahriar Ghammamy, 2012. Structural Properties, Natural Bond Orbital, Theory Functional Calculations (DFT) and Energies for the Halorganic Compounds Current World Environment, 7(2): 221-226.
- Jursic, B.S., 2000. A B3LYP hybrid density functional theory study of structural properties,and energies heats of formation for silicon–hydroge compounds, Journal of Molecular Structure (Theochem), 497: 65-73.
- Gardiner, D.J., 1989. Practical Raman spectroscopy. Springer-Verlag. ISBN 978-0- 387-50254-0.
- Abou-Deif, M.H., M.A. Rashed, M.A.A. Sallam, E.A.H. Mostafa and W.A. Ramadan, 2013, Characterization of Twenty Wheat Varieties by ISSR Markers, Middle-East Journal of Scientific Research, 15(2): 168-175.
- Kabiru Jinjiri Ringim, 2013. Understanding of Account Holder in Conventional Bank toward Islamic Banking Products, Middle-East Journal of Scientific Research, 15(2): 176-183.
- Muhammad Azam, Sallahuddin Hassan and Khairuzzaman, 2013. Corruption, Workers Remittances, Fdi and Economic Growth in Five South and South East Asian Countries: A Panel Data Approach Middle-East Journal of Scientific Research, 15(2): 184-190.
- Lewis, D. F. V., C. Ioannides, and Parke, D. V. 1994. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: computer analysis of structure- activity relationships. Xenobiotica, 24: 401-408.
Recommended for publication by Prof. S.M. Igoumnov
Fluorine Notes, 2014, 96, 5-6
