Received: October, 2012
Fluorine Notes, 2014, 95, 1-2
Chemistry and Technology of Polyfluorinated Organic Compounds Based on New Aggression Resistant Catalysts
Vladimir Y. Zakharov
State Educational Institution of Higher Professional Education
"Vyatka State University", 610000,
Kirov, Moskovskaya str. 36, Russia
e-mail: zakhar.05@mail.ru
Abstract: Scientific principles of selection of effective catalysts of direct gas-phase fluorination of organic compounds have been developed. The use of created catalysts improve existing and allow to develop the new technologies of polyfluorinated organic products.
Keywords: Organofluorine compounds, fluorine, cobolt trifluoride, isomerization, heterogeneous catalyst, catalytic fluorination.
1. Introduction
As materials possessing a number of findunique properties fluororganic compounds find wide application in different fields of technics. Fluoroplasts, fluorelastomer, special fluorinated lubricants and liquids are irreplaceable while working with aggressive media under conditions – using of those materials, particularly, led to revolution in the atomic industry technology, in airspace and reaction (jet) technics, as well as in high-speed aviation.
Special physical and chemical properties of fluorochlororganic liquids (e.g. chladones and freons) provided/provoked rapid development of refrigerating technics.
Compositions using polyfluorinated organic compounds (“light water”) form the base for modern flame extinguishing means.
Aqueous emulsions of a number of fluororganic liquids, in particular perfluorodecaline are the effective carriers of oxygen; this creates perspectives for their usage as blood substituents in medicine. High biological activity of a number of polyfluorinated organic compounds is being used in both medicine (cancer fighting medicines) and agriculture (pesticides). Even that, far from full list of application fields for fluororganic products allows appreciating of their applied meaning. A wide range of perfluorinated products can be obtained by fluorination of polyfluorinated organic compounds using elemental fluorine. At the same timestudies on interaction of fluorine with organic compounds are held by non-selective, explosive nature of that process caused by its high exothermicity.
Application of heterogenic catalysts is one of the ways to increase selectivity of direct fluorination; however, it should be noted, that though the achieved level of catalytic fluorination technique allows singnificant increase of selectivity compare to homogenious gas-phase process, nevertheless it is as a rule characterized by high destruction of substrate, and sometimes by contraversioal, unrepeatable results. The developed (known) catalysts proved to be not effective enough for usage on enlatrged, industrial scale, and catalytic fluorination method, as a consequence, did not gain practical ground of wide range. We also should note, that high reactivity of fluorine dramatically narrows the range of materials applicable for preparation of stable catalysts. All of that outlines the actuality of studies aimed at creation of effective aggression resostant heterogenic contacts and development of direct fluorination technique based on them, which backs the opportunity of its wide range practical usage. Solving this important task was held in our opinion by the absence of new rational approaches to the selection of direct organic compounds gas-phase fluorination catalysts.
Selectivity and rate/speed of a whole class of practically important thermal transformations of polyfluorinated organic compounds (oxidation, dechlorination with hydrogen, isomerization etc.) also can be increased when using effective catalysts, however, practical application of catalysis for solving the actual tasks of fluorine products technology regarding decreasing the materials consumtion and energy capacity, environmental pollution, synthesis of new unique materials is obviously not enough. When creating stable, commercial catalysts of thermal transformations of polyfluorinated organic compounds a high aggressivity of a reaction medium is also one of the problems, that results in premature destruction of heterogenic contacts. All that stipulates the actuality of studies in the field of selection of effective aggression resistant catalysts of thermal transformations of polyfluorinated organic compounds.
In the first part of the present work the scientific priciples of selection of effective catalysts of organic compounds direct gas-phase fluorination are developed and for the first time the opportunity of sharp increase of selectivity and rate of fluorination by undiluted fluorine at directed change of nature of catalytic composition is experimatally shown. The selected universal aggression resistant catalytic composition NiF2/О±-Al2O3 allowed not only carrying out the synthesis of a number of practically important perfluorocarbonic products with quantitive yield (2-perfluoromethylperfluoropentane, 3-perfluoisopropyl-2-perfluoromethylperfluoropentane, perfluoroethane, perfluorobutane, pentafluorochloroethane etc.), but also for the first time introduce into industrial practice the technology of direct catalytic fluorination (the production of perfluorodimethylperfluorocyclohexane, the purification of perfluorodecaline, B-I, M-I liquids, lubricants, regeneration of fluorocarbonic liquids etc.)
In the second part of the work based on the systematical study of wide class of thermal gas-phase transformations of polyfluorinated organic compounds it was has been stated, that О±-AI2O3 is a universal aggression resistant carrier for the catalysts of these processes. A number of new effective heterogeneous contacts Based on О±-AI2O3especially for the catalysis of 1,2-dichlorohexafluoropropane and 1,2-difluorotetrachloroethane dechlorination using hydrogen, as well as oxidation of tetrafluoroethylene.
As a result of studying the kinetic particularities of thermal gas-phase reactions of polyflorinated organic compounds it has been stated, that dehydrated activated carbon is an effective aggression resistant catalyst of nucleophilic type and it can be widely used even in unmodified form, especially, for catalysis of hexafluoropropyleneoxide’s isomerization, destruction of carbonyldifluoride oligomers, perfluorinated polyperoxides and ω-chlorperfluoroalkylfluorosulphates, hydrolysis and hydrofluorination of perfluoroisobutylene.
Application of created thermal aggressive resistant catalysts allowed improving existing and developing new resource saving, more ecologically pure technologies of a number of polyfluorinated organic products (hexafluoropropylene, 1,2-difluorodichloroethylene, carbonyldifluoride, perfluoropolyoxamethylenacetylfluorides etc.)
2. Direct Catalytic Fluorination Of Polyfluorinated Organic Compounds.
2.1. Analytical Review
Chemism of direct fluorination of organic compounds, which according to /1-3/ goes along radical type, is to a great extent determined by thermodynamical particularities of its certain stages:
F2 → 2F• +155  kJ/mole
RH+F• → R•+HF – 140  kJ/mole
R• + F2 → RF+F• – 285  kJ/mole
These particularities lie in rather low energy of dissociation of molecular fluorine (155 В kJ/mole) and high heat which is isolating during fluorination (more than 420 В kJ/mole of fluorine). If we take into account, that the energy of C-C bond in the molecules of hydrocarbons consists approximately 330В kJ/mole it is geeting clear, why the branching of chains with destruction of carbon frame of subsrate is quite typical for direct fluorination.
The dilution of fluorine with inert gases /4-11/, the dissolution of organic substrate in the inert to fluorine solvent /12-18/, the cooling of reaction mixtures /19-32/, using of reactors with special constructions /33-37/ - jet one, with zone heating, as awell as controlled fluorination in solid phase /38-51/ are appllied to increase the selectivity of direct fluorination.
Along with direct fluorination electrical and chemical /52-58/ and metal and fluoride methods /59-70/ have been developing; the last one is based on using metal fluorides mainly cobalt in the state of their higher valency as fluorinating agents.
Using these milder compare to elemental fluorine fluorinating agents allows providing firm control of fluorination temperature, which is especially important when conducting a process on an enlarged scale.That is why the metal fluoride method inspite of high material and energy consumption as well as low productivity formed the base of existing fluorinating technologyfor organic compounds.
One of the ways to increase selectivity of fluorination is conducting interaction of elemental fluorine and organic substrate in the layer of catalyst /71/. During WWII that method formed the base for the first production of strategically important fluorocarbons /72/. As it follows from /73-75/ the dispersing of heat isolating during fluorination is the main function of catalyst (packing), that allows escaping local overheatings, lowering averagemass temperature of reaction gases and then by doing so decrease the input of destruction of initial raw materials, intermediate and end products. Due to that the wide use of metal catalysts – copper, silver, nickel of high thermal conductivity is becoming understandable. /2,72-79/.
For the first time the use of metal catalyst at fluorination according to /2/ was described by by Fredenhagen and Kadenbachen: fluorine mixed with nitrogen (mole ratio 1: 1) was being introduced into glass reactor (diameter – 6cm, length – 40cm) by perfluorinated copper tube wrapped in a few layers of copper net. The reactor was being turned at such a rate that the organic liquid in the bottom dispersed in the form of film along its walls.
Later Bigelow used the reactor of analogous construction (diameter -2,8cm, length – 75cm, input of fluorine and nitrogen at a mole ratio 1:1, 6 along perfluorinated copper tube wrapped with copper net) for fluorination of tetrachloroethylene and hexachloroethane. The substrate ws being put on the bottom of reactor and also closed with the copper net; yield of 1,2-difluorotetrachloroethane – the product of selective attachment of fluorineachieved 20% at that.
In /2,73,80/ the data on gas-phase direct fluorination of ethane, methane, ethyl chloride, 1,2-dichloropropane, benzole, toluene, benzotrifluoride and acetone in the layer of dense nozzle made of copper net or copper wire rolls is listed. Besides the main product – tetrafluoromethane, hexa-, penta-, tetra- and trifluoroethane have been obtained fluorinating ethane; the formation of di- and monofluoromethane was not observed. During fluorination of methane, besides tetra-, tri-, di and monofluoromethane the formation of hexafluoroethane and octafluoropropanehas been registered, which shows the association of intermediate radicals /2,73/.
During fluorination of ethyl chloride 1,1-dichloroethane and 1,1-dichlorine-2,2-difluoroethane have been found in the reaction products, besides tetrafluoromethane and products of selective substitution of hydrogen atoms for fluorine. This result points out, that during fluorination chlorination occurs; it takes place either due to formation of fluorine chloride which is a strong chlorinating agent or as a consequence of substarte’s interaction with isolating free chlorine /73/.
The same results were obtained during gas-phase fluorination of trichloroethylene over ground copper /2/ - tetrachlorodifluoroethane is one of the main products of reaction.According to /80/ trichloropentafluoropropane and tetrachlorotetrafluoropropane are one of the main products of reaction of direct fluorination of 1,2-dichloropropane in the layer of nozzle made of copper wire at 429-470 K; the yield of 1,2-dichlorohexafluoropropane – the product of selective substitution of hydrogen atoms for fluorine is 15-25% under these conditions.
Destruction of carbon frame and polymerization are rather typical for direct gas-phase fluorination in the layer of copper nozzle; thus, CF4, C2F6, C3F8, C4F10, C5F12, C6F12, C6HF11 and C12F22 /73/ have been isolated from benzene fluorination products. The yield of perfluorodecaline during tetralyne (620 – 650K) fluorination over silver plated copper filings amounted only 15% /81/. Only insignificant amounts of perfluorodecaline form during fluorination of α-methylnaphtalene over silverplated copper wire /73/. During fluorination of benzotrifluoride under the same conditions significant amounts of destruction products and partial fluorination products, including perfluorocyclohexane and perfluoromethylmonohydroperfluorocyclohexane are formed besides perfluoromethylperfluorocyclohexane. /73/.
At the same time /72/ it is being noted, that modifying of copper filings using silver (in rare cases – gold) allows achieving significant increase of selective exhaustive fluorination products yield (Table 1).
Also 2,2,3-trimethylbutane, 2,2,4-trimethylpentane, cetane, toluene, mezytylene, reten and christen /72/ were subject to direct fluorination using that method. The characteristics of products of their selective fluorination are listed in /82/.
In /73/ the following conditions are formulated to achieve maximum yield of fluorocarbons at direct fluorination:
- application of catalyst – silver on metallic nozzle;
- carrying out the process at the excess of fluorine;
- keeping the temperature within the range of 410-600 K (at temperature below 410 K organic compounds condensate at nozzle and destruct in fluorine, and at temperature over 600 K the nozzle cripples);
- fluorine and organic substarte vapour must mix up in the catalyst’s layer.
Table 1. Fluorination of Hydrocarbons Over Silver Plated Copper Nozzle /72/.
|
Initial Compound |
Reaction Temperature, K |
Yield of Selective Exhaustive Fluorination Product, % of Theoretical One |
|
C6H6 |
538 |
58 |
|
n-C7H16 |
408 |
62 |
|
C6H5CF3 |
473 |
85 |
|
C6H4(CF3)2 |
473 |
87 |
|
Antracene |
573 |
43 |
|
Naphthenic oil |
563 |
15 |
|
Aromatic oil |
573 |
19 |
According to /2/ the copper fluoride covering copper nozzle is not a fluorinating agent as direct
experiments show – in the authors’ opinion, this fact reduces the nozzle function to the elimination
of local overheatings and excludes the chance of its catalytic influence. On the contrary, silver
difluoride is an effective fluorinating agent /2,61/. In this connection the authors think /72/,
that silver fluoride is the main fluorinating agent when using copper nozzle modified by silver,
and the role of elemental fluorine is reduced to regeneration of that fluoride.
At the same time authors /74, 75/ come to the contrary conclusions and think, that modification of copper nozzle using silver doesn’t have any significant influence on its catalytic properties. Comparative data on benzene fluorination at 538K at different metallic catalysts /74/ are listed below (Table 2).
Analyzing the listed information we can see, that catalytic influence of most metal nozzles is practically the same; it can also be seen, that silver plating doesn’t influence catalytic properties of copper filings.
Table 2. Direct Catalytic Fluorination Of Benzene.
|
Catalyst |
Yield of C6F12, % Of Theoretical One |
|
Copper Filings |
29 |
|
Silver Plated Copper Filings |
26 |
|
Gols Plated Copper Filings |
35 |
|
Nickel Plated Copper Filings |
23 |
|
Cobalt Covered Copper Filings |
28 |
|
Brass Filings |
24 |
|
Amalgamated Copper Filings |
10 |
|
Copper Wire Plated with Rhodium |
17 |
|
Chromium Plated Copper Filings |
7 |
In /2/ it is noted, that in some cases modified catalysts proved to be less effective, than unplated copper and nickel fraction, and we make a conclusion, that the main function of metal nozzle is in dispersing of reaction heat, as a consequnce of which the fluorination goes at moderate temperatures. It is also being noted /2/, that the form of nozzle (net, filing, fraction) is less important, that the nature of metal surface.
Cobalt trifluoride is also described as a catalyst of direct fluorination of aromatic compounds /83-85/. The processing of О±-methylnaphtalene using elemental fluorine at 633 K in the layer of cobalt trifluoride is mainly accompanied by destruction; the yield of perfluoromethylperfluorodecaline amounted to 11%. The fluorination of perchlorocyclopentadien under the same conditions results in formation of pentachloropentafluorocyclopentane with the yield of 70% /83/.
We are informed about obtaining of liquid fluorocarbons by exhaustive fluorination of pentane, hexane, toluene, metha-xylol, tetralyne, О±-methylnaphthalene /84, 85/, alkyladamantanes, adamantancarboxylic acids, derivatives of bicycloheptane, bicyclononane /86/; the substrate is evaporated and filtered through fluidizated layer of cobalt trifluoride at 600-725 K over elemental fluorine.
Partial fluorination of hydrocarbons by fluorides of silver, manganese, sulphur or antimony preceeds the stage of fluorination using elemental fluorine /86/. Elecrochemical fluorination of cycloalkenes or their partly substituted fluorine derivatives in anhydrous hydrogen fluoride over cobalt fluorides, nickel and manganese fluorides is described in /56/.
The fluorination of organic compounds on the particles of alkali metal fluoride dispersed in the flow of inert gas is described in /87,88/. Authors /87/ mixed organic substrate with sodium fluoride, carefully triturated it, sprayed in the flow of nitrogen and introduced for fluorination; aromatic compounds (benzene toluene, naphthalene), aliphatic and cycloaliphatic hydrocarbons (for example, hexane, dodecan), polyethylene, substituted hydrocarbons (nitrobenzene, benzoic acid), and also some dibasic acids were used as substrate.A significant input of association of intermediate radicals is noticed at low-temperature fluorination of aromatic compounds on sodium fluoride in /87/. Thus, the mixture of compounds of average molecular weight of 670 was obtained at 253 K using fluorination of benzene by fluorine, diluted nitrogen (rate 1 : 10), products of average molecular weight of about 1000 were obtained under analogous conditions out of toluene.
The authors /88/ prepared spray of sodium fluoride in the flow of helium beforehand, mixed it with the flow of helium saturated with substare vapours – pentane, cyclohexane or dioxane, and directed them into precooled fluorinating reactor.Fluorinating cyclohexane, for example, at 210 K at at rate fluorine : substrate equal to 1 : 1 and at tenfold dilution of fluorine with helium monofluorocyclohexane of 26% yield, difluorocyclohexane (20%) as well as other polyfluorinated products were obtained. Authors /88/ have used the reactor equipped with multisection cooling allowing keeping up the certain temperature in each zone of reaction zone for controlling the selectivity of fluorination. Direct fluorination of nitrogen-, oxygen-, and sulphur containing organic compounds at alkali metals fluorides is described in /89-97/.
The catalytic fluorination of 2-hydroperfluoropropane at copper, silver, gold, mercury, cobalt, platinum, their fluorides or alloies with obtaining of octafluoropropane is described in /98-100/. Initially the starting material – hexafluoropropylene is being processed with anhydrous hydrogen fluoride at 370-570 K over chrome oxifluoride. 2-hydroperfluoropropane forming at that is put into the layer of catalyst at 370-620 K together with elemental fluorine (fluorine excess is (5-30%), diluted nitrogen (1 : 1). The yield of octafluoropropane at 510-520 K on silver plated copper filings preliminary treated with fluorine amounted to almost 95% of theoretical one (onto hexafluoropropylene reacted).
The obtaining of octafluoropropane using direct fluorination of propane or propylene at 330-510 K over fused porous aluminium oxide is described in /101, 102/. Fluorine is introduced into the inner part of the tube made of fused aluminium oxide, through the pores of which it is migrating into the outer part, where the carbon is introduced to. At that the yield of octafluoropropane amounts to 10-85% of the one possible in theory /101/. Metallic copper and nickel /102, 103/ are described as porous element; porous tube, thourh which fluorine is migrating can be impregnated with catalytically active compounds /104/.
Cobalt /73/, aluminium /105/, antimony and titanium /106/ fluorides as well as chloric iron are used as catalysts of selective substitution of haloid atoms (mainly, fluorine) for fluorine during direct fluorination of organic compounds.Thus, authors /105/ used aluminium trifluoride (in the form of crystals of 500Зє size) preliminary contacted with oxygen at 670-730 K /108/as a catalyst; the fluorination was carried out within the temperature range of 380 K (at the entry of reactor) to 630 K (at the exit of reactor) at mole ratio chlorofluorocarbon : fluorine equal to 1 : 1,7 and contacting period equal to 125 sec. Application of aluminium fluoride allowed obtaining of perfluoropropane of 81% yield out of 1,2,2-trichloroperfluoropropane, and perfluorobutane (yield is 76%) out of 2,3,3-trichloroperfluorobutane.
The work of Bigelow and co-workers, in which acetone was used as a substarte, and copper net – as a catalyst is considered a first successful experiment on direct gas-phase fluorination of oxygen containing organic compounds. Reaction gases contained significant amounts of destruction products – tetrafluoromethane, carbonyldifluoride and perfluoroacetylfluoride besides perfluoroacetone and products of incomplete fluorination of acetone. The yield of perfluoroacetone amounted to 10% at a rate of fluorine : acetone : nitrogen equal to 6:1:19 and starting temperature in reactor of 333 K. Fluorination of acetone with more concentrated fluorine (fluorine : nitrogen = 1 : 1) at temperature of 473 K in reactor filled with gold plated or platinum plated copper filings is characterized by increasing of yield of destruction products. Octafluorobutanone -2 /2/ is obtained by fluorination of methylethylketone with the yield of 10% under analogous conditions.
According to /73/ Cady and co-workers carried out direct fluorination of alcohols for the first time; carbonyl fluoride and trimethylhypofluorite were the main products of methyl alcohol fluorination with diluted alcohol at 450 K over silver plated cooper band.In /109-120/ you can find information on obtaining trifluoromethylhypofluorite, difluoromethylen-bis-hypofluorite and perfluoroalkylhypofluorites by selective attachment of fluorine along double bond carbon-oxygen; alkali metals fluorides (especially – cesium), active at even 195 K and below and silver and nickel fluorides are effective catalysts of this process.
The results on direct fluorination of nitrogen containing organic compounds are listed in /121, 122/. Tetrafluoromethane was the main product of methylamine fluorination at 370 K (rate fluorine : amine was equal to 5 : 1 : 15) in the layer of copper fraction; at that CF3NF2, CF3CF2NF2 and (CF3)2NF /121/ also are formed. The composition of reaction products shows significant destruction of raw materials and further partial association of forming radicals.
Fluorination of malononitrile at 523 K in the layer of copper fraction (rate fluorine : malononitrile : nitrogen equaled to 3: 1 : 9) is characterized by formation of tetrafluoromethane (13 mass.%), octafluoropropane (30 mass. %), and also products of destructive fluorination and association of forming radicals /122/.
A number of sulphur-organic compounds had undergone direct catalytic fluorination; thus, in /71/ we are being informed, that interaction of methylmerkaptane with elemental fluorine at 437 K in the layer of silver modified copper net is accompanied by forming (with small yield) of hydrogen atoms’ selective substitution products - CF3SF5 (15%) и CF3SHF4 (15%).
Thus, direct gas-phasefluorination of organic compounds is charactirezed as a rule, by significant input of destructive fluorination with breakage of C-C bonds in the molecule of substrate, which is due to high energy isolating during elemental stages of that exothermal process.Heterogenic catalysts – mainly metals (copper as a rule), which in a number of cases are being modified (in most cases – using silver) are used for decrease of destruction at gas-phase fluorination. Metallic catalysts are chosen because of their high thermal conductivity, it provides for effective heat removing from the areas of local overheating and thereafter decreasing of destruction.
At the same time it should be noted, that though catalytic fluorination allows significant increasing of process selectivity (compare to homogenious gas-phase fluorination), nevertheless it is characterized as a rule, by relatively high destruction of the raw materials, and sometimes controversial, unrepeatable results. The developed catalysts proved to be insufficiently effective for their use in on enlarged industrial scale, and as a consequence the catalytic fluorination method did not receive wide practical application.This fact outlines the actuality of studies aimed at creation of effective catalysts and development of direct fluorination technology based on them to the level providing the opportunity of its practical use on a large scale.In our opinion, the resolving of this important task was held by the absence of new, rational approaches to the creation of effective catalysts of direct gas-phase fluorination of organic compounds.
2.2 Scientific Foundations for Selection of Effective Aggression Resistant Catalysts Of Organic Compounds’ Direct Gas-Phase Fluorination
The following considerations are laid as a base of developing by us approach for selection of effective catalysts of fluorination of organic compounds.
1. Gas-phase interaction of organic compounds with fluorine leads to formation of intermediate adducts possessing excessive energy. These exited adducts undergo either spontaneous destruction or transfer into main state due to clash with molecules of gas phase or surface.
2. We can suppose, that improving the selectivity of interaction of organic compounds and fluorine will be achieved at localization of elemental acts of fluorination on the surface. In this case the probability of timely removal of execessive energy of exited adducts along the bonds with elements of nozzle surface, which acts as averaging “thermal reservoir” sharply increases. At that the destruction of exited adducts decreases, but not the thermal destruction (for example, because of strong heating of reaction gases).
3. To localize the elemental acts on the surface not only has the catalyst to intensively sorbate molecular fluorine, e. g. to possess well-developed related to fluorine surface, but also activate it.
Only in this case kinetic background for localization of the process directly on the surface can appear.
Thus, the preference at choosing the fluorination catalyst must be given to the contacts with developed surface modified using activating fluorine additives. The presence of developed surface leads to improving of selectivity also due to the breakage of chains of radical branching process. According to that the low effectiveness of metallic catalysts is becoming clear, their exterior surface is quite small.
4. High aggression of the medium is a specific feature of direct fluorination; it applies additional requirements to the stability of catalysts.
The main principal feature of this new approach to the creation of effective catalysts of direct fluorination is concluded in the fact, that we can suppose the possibility of sharp increase in selectivity due to localization of fluorination acts on the surface, but not due decreasing or averaging of reaction gases temperature (removing of local overheatings) when using thermal conductive nozzle – catalyst, as it had been done before.
Actually, the destruction of raw materials is often observed /19, 87, 88/ at fluorination under regulated mild conditions (273 K and below), when thermal breakage of molecular bonds is unlikely to happen – this effect can be explained only by the decomposition of energetically exited adducts /123,124/. Energetic branching of chains is quite typical for low-temperature gas-phase fluorination and defines, in particular, kinetic particularities of its passing /125-136/. Thus, authors /125-129/ explain the significant decreasing of fluorination rate at transfer from difluoromethane to trifluoromethane by the possibility of chains branching at fluorination of CH2F2 at account of decomposition of exited molecule of forming CHF3:
CH2F2 + F• → CHF2 + HF
CHF2 + F2 → CHF3* + F•
CHF3* в†’ :CF2 + HF
:CF2 + F2 → CF3• + F•
During fluorination of trifluoromethane such an opportunity is absent, that causes abrupt decrease of the process rate.In this connection the observed in /132-133/ high fluorination rate of dichloromethane and fluorochloromethane – there is also a chance of energetic branching of chains during decomposition of energy-saturated intermediate adducts.
In this connection it is interesting, that difluoromethane fluorination rate significantly decreases, and the yield of trifluoromethane is increasing at highering of parcial pressure of inert gas; this effect can be explained /134/ by deactivation of exited molecules of trifluormethane forming at clash with molecules of inert gas.
The ratio of energetically exited molecules at fluorination decomposition rate and thermally active molecules decomposition rate is clearly characterized by the data on selectivity of fluorochlormethane fluorination /133/ – decomposition of difluorochloromethane forming passes only with detaching of anhydrous hydrogen fluoride, but not hydrogen chloride, which is typical for thermally active molecule of difluorochloromethane, for example, at its pyrolysis /137/.
Decomposition rate along exited C-F bond is significantlt higher than redistribution rate of excessive energy along non-exited bonds, that causes unusual selectivity of destruction of difluorochloromethane forming /133/. These results speak about/demonstrate/#ify the impact of decomposition of forming exited adducts on selectivity of fluorination of organic compounds using elemental fluorine.
Below you will find the principal kinetic scheme for the discussed above model of catalytic fluorination, analysis of which clearly illustrates the demands to effective catalyst1:
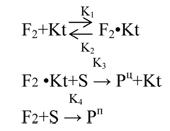
1The scheme has got common character and doesn’t focus on, for example, elementary stages, their radical mechanism, and describes only main patterns of fluorination characterized by the presence of competeing processes in gaseous phase and at the surface.
At that, it is being supposed, that interaction between adsorbed fluorine (F2•Kt) and substarte (S) at the surface of catalyst is accompanied by forming of only target products (Pц) at a rate characterized by effective K3; the fluorination with participation of gasiform fluorine (F2), which is characterized by K4 constant leads to forming of only by-products (Pц).
The analysis of this scheme shows, that reciprocal value of fluorination selectivity (I/О±), is described by the equation:
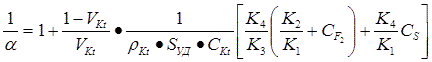
Driving from that it is seen, that selective fluorination is provoked by the increase of share of reaction volume, filled with catalyst (VKt), the increase of its specific surface (SСѓРґ) and surface concentration of active centres (CKt) as well as increased sorption ability towards molecular fluorine presented at contact (K2/K1) and its activating influence onto adsorbed fluorine (K3). On the contrary, selectivity is decreasing at gas-phase fluorination input increasing (K4), as well as concentrations of fluorine (CF2) and substrate (Cs).
It should be noted, that demands to effective catalyst of fluorination ensuing from model under consideration are rather universal; for example, the activation of fluorine may include its dissociation on the surface forming atomic fluorine, which reactivity is increased to a wide range of substrates’classes (but not to all of them). The selection of catalyst component activating fluorine most effectively is a first stage in determining the composition of optimal catalytic composition.
Gas-phase interaction of carbon and fluorine oxides forming trifluoromethylhypofluorite (THF) – the effective initiator and reagent to synthesize a wide range of fluororganic compounds was studied as a model reaction /138 – 140/.
Catalytic Fluorination of Carbon Oxide
Gas-phase fluorination of carbonoxide includes two stages:
CO + F2= COF2 –525  kJ/mole
COF2 + F2= CF3OF –135  kJ/mole
The rate of second stage of this process – interaction of carbonyl difluoride and fluorine can be significantly increased when using catalyst or initiator /112-118, 141-142/.
Studying the catalytic properties of silver, nickel, cobalt, copper, manganese, iron, cesium, potassium, lithium, barium, strontium, calcium and magnesium fluorides applied on Оі-AI2O3in equimolar quantities using impregnation method proved, that activity of those contats is unequal and it is determined by the nature of metal fluoride applied. As you can see from the Pic. 1 the activity correlates with ionization potential of corresponding cation.This result is understandable if we take into account, that the character of fluorine molecule is pronounced as electron-seeking. Partial transfer of electrone from cation of ionization low potential onto Пѓ-looseningorbital of adsorbed fluorine molecule eases the dissociation of halogen forming active atomic fluorine /143/, that causes increase of reaction rate.
Applied nickel, silver and cesium fluorides are of most high catalytic activity as it follows from Pic. 1.
In /144-147/ it is being noted, that nickel is an effective catalyst of dissociation of molecular fluorine into atoms. Adducts of interaction of nickel and fluorine are described by the formula NiFx, where 2≤x≤3; their IR-spectra coinside with spectra of nickel trifluoride /142, 145, 148/. Adducts of interaction of nickel chloride and fluorine are also described by the formula of NiF2,5 /149/.
Obtained in /139/ kinetics data on direct fluorination of carbonyl difluoride on nickel difluoride agrees with process scheme including atomization of fluorine on catalyst (reaction order on fluorine equals to 1, 5). According to estimates listed in /139/ the rate of fluorine dissociation at nickel difluoride increases by more than 5 times (compare to non-catlytic dissociation).Atomization of fluorine on the surface of lowest fluorides of nickel and silver accompanied by forming of highest fluorides /149/ apparently determines their catalytic activity in direct fluorination of carbon oxide.
Catalytic influence of cesium fluoride at fluorination of carbonyl fluoride – the intermediate product in the synthesis of THF is caused as follows from /112-116/ by activation of substrate (and not the fluorine) forming corresponding cesium alcoxide; this result was obtained when carrying out the reaction under rather mild conditions – 200 K and below.
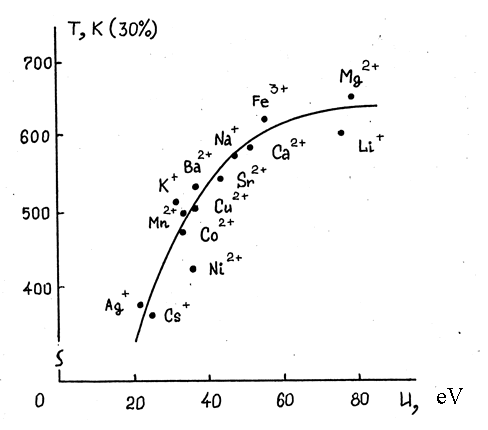
Picture 1.Correlation of applied (coated) metal fluorides’activity in catalytic fluorination of carbon oxide and ionization potentials (u, eV) of corresponding cations (T temperature was taken as a measure of activity, at that T the concentration of trifluoromethylhypofluorite in reaction gases reached 30 volume % at a rate of reagents’ supply equal to 1800 hour-1 and mole ratio equal to F2:CO=2).
At the same time it is being noted in /150, 151/, that alkali metals’ fluoroalcoxides, in particular – cesium, are being destroyed at temperature over 470 K and, thus, are unstable under studied conditions of THF synthesis (up to 650 K). That is why the catalytic influence of cesium fluoride at hightemperature fluorination of carbon oxide till THF can be caused by fluorine activation.
High-temperature catalytic fluorination of carbon oxide, including the one at cesium fluoride applied is characterized, as the analysis of reaction gases proved/showed, by forming of significant amounts of bistrifluoromethyl peroxide /152/, what, in author’s opinion /115/, testiifies in favour of fluorination radical mechanism with atomic fluorine participating.
It should be noted, that electrochemical fluorination of carbon oxide, carbonyldichloride and carbonyldifluoride under mild conditions passing with participation of atomic fluorine is characterized by high yield of THF /153-154/. That result points out high activity of fluorine atoms at synthesis of THF and witnesses in favour of offered fluorine activation mechanism at active component of catalyst during fluorination of carbonoxide.
It is typical for the catalysts prepared by mechanical mixing (thorough pestling in agate mortar) of metal fluoride components and γ-AI2O3, to be significantly less active than the catalysts obtained by method of coating (application) (impregnation). Apparently, that result is caused by different level of dispersity of active component and indicates significant influence of ctalysts’ preparation methods on their efficiency. It is interesting, that coated (applied) catalysts of NiF2/γ-AI2O3, obtained using nickel chloride, nitrate and sulfate (at preliminary fluorination of catalysts these salts applied on γ-AI2O3, transfer to nickel difluoride) are approximately equally acitive.
We should also note, that known metal catalysts – dense nozzle made of thin copper wire and rollers of nickel net were pvirtually inactive under analogous conditions. Apparently, that is a consequence of their low specific surface. Specific surface of metal catalysts – dense nozzle made of copper wire spirales and rollers mde of nickel net accounts to 2,8 x 10-3m2 and 3,5 x 10-3m2per 1 ml of catalyst respectively. For comparison the specific surface of waste/used catalysts based on γ-AI2O3is 15-20 m2/ml.
Choosing the carrier was the following stage of determining the optimal composition of direct fluorination. The X-ray phase study of used/waste catalysts based on γ-AI2O3and determining of their chemical composition proved, that total fluorination of contacts occurred during reaction forming phase of aluminium fluoride. That phase transfer is accompanied by decrease of mechanical stability/resistance/ruggness of catalysts, that led to gradual decomposition of their granules and to increase of reactor’s resistance.
An interesting observation has been made during studying of catalytic propertie of nickel oxide applied on О±-AI2O3; not only was that catalyst of high activity during carbon oxide fluorination obtaining THF, but also has it preserved its initial strength and granulometric composition:
|
Catalyst |
Operating Conditions |
Overall fraction weight, % C grains size less than1, mm |
||||
|
0,5 |
2,0 |
3,0 |
5,0 |
7,0 |
||
|
NiO/Оі-AI2O3 |
553 K, 24 hours |
12,6 |
45,8 |
73,2 |
91,9 |
100,0 |
|
NiO/О±-AI2O3 |
623 K, 200 hours |
0,5 |
0,8 |
1,0 |
2,9 |
100,0 |
1Size of initial grains – 5-7 mm.
From the listed data we can see, that only 8,1% of grains of catalyst based on Оі-AI2O3 have remained unaltered in their size after operating for 24 hours at 553K; all the others had been destroyed.The catalyst based on О±-AI2O3, operated under more severe conditions (623 K, 200 hours) had remained its granulometric composition virtually unaltered.
X-ray-phase analysis of the catalyst NiО/α-AI2O3 had revealed, that during operation or preliminary fluorination the coated (applied) oxide fully transformed/transferred into nickel fluoride, while the intensity of lines responding to the carrier – α-AI2O3stays the same. The presence of saturation lines (of small intensity) in spectra responding to the aluminium fluoride provides a basis for making an assumption, that during preliminary fluorine treatment (using fluorine) and operation the α-AI2O3is being coated with thin aluminium fluoride film, which prevents it from further fluorination and thus, from mechanical decay. Thus, used catalyst is a nickel fluoride applied (coated) onto α-AI2O3, which is coated with aluminium fluoride. To keep it short this contact is referred to as NiF2/α-Al2O3.
Here we shall note, that exclusively high stability/ resistance of О±-AI2O3amid fluorinating media at increased temperatures, that we had discovered during our work, allowed using of that carrier (as will be shown below) to prepare effective catalysts of a wide range of fluoroorganic compounds reactions..
In the reaction of carbonyl difluoride’s fluorination the tests of GIAP-3-6H commercial catalyst, which is also a nickel oxide applied on α-AI2O had confirmed a high efficiency of that catalytic composition (Table 3) and allowed obtaining of reaction gases containing THF at a point exceeding 95% of volume.
Table 3. Direct Fluorination of Carbonyl Difluoride at GIAP-3-6H (volume rate of reagents’ input - 100 hour-1; mole ratio substrate:fluorine=1).
|
# |
Temperature, K |
Composition1 of Reaction Gases, % volume |
|||
|
F2+CO |
CF2O |
(CF3O)2 |
CF3OF |
||
|
1 |
448 |
2,6 |
1,7 |
0,4 |
95,3 |
|
2 |
473 |
2,2 |
1,4 |
0,2 |
96,2 |
|
3 |
498 |
1,8 |
1,1 |
<0,1 |
97,1 |
1Content of tetrafluoromethane and carbon dioxide in the reaction gases below 0,1 volume %.
We shall note, that cesium and silver fluoride applied on α-AI2O3, are also initially catalytically highly active in THF synthesis based on fluorination of carbon oxide; in this case as well the carrier α-AI2O3remains its high mechanical stability even after prolonged use under rather severe conditions. At the same time lowering of activity in time is typical for these catalysts, which is caused by gradual layering off and carrying away of active components – metal fluorides; the latter leads to increase of catalysts’ layer’s resistance and plugging in reactor.
Besides that, cesium fluorides and especially silver fluorides are expensive and hard to find.Due to that later the main part of our attention was devoted to studying of catalytic properties of active, stable and available NiF2/О±-AI2O3, as well as their comparison to the properties of inert nozzles, for example, melted calcium fluoride, non-modified О±-AI2O3) and known metal catalysts.
Under fluorinated addmixtures are a complex mixture of polyfluorinated unsaturated and hydrogen containing compounds. We have conducted a study of model reactions of direct catalytic fluorination of a wide range of unsaturated and hydrogen containing individual polyfluorinated compounds to select effective catalysts of their direct fluorination. However, in a number of cases studying the model reactions of direct fluorination had been of independent practical ineterst.
2.3 Catalytic Gas-phase Fluorination of Double Bond Polyfluorinated Compounds
2.3.1 Perfluorononenes’ Fluorination
We have studied the gas-phase fluorination of perfluorononenes (PFN) as a model reaction to estimate the effect of fluorine activation by the surface on rate and selectivity of its attachment by double bond.
Perfluorononenes are formed with high yield at anionic oligomerization of hexafluoropropylene (155-162):
|
|
|
3-perfluoroisopropyl-2-perfluoromethylperfluoropentene-2 (PFEN-1) 60 mass.% |
|
|
3-perfluoroisopropyl-4-perfluoromethylperfluoropentene-2 (PFEN-2) 30 mass.% |
|
|
|
2,4-perfluorodimethylperfluoroheptene-3 (PFEN-3) 5 mass.% |

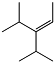
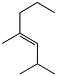
Perfluorononanes (PFAN) – the products of selective fluorinations of PFEN, they are perspective dielectrics, coolants and thermal resistant solvent.
Information on direct gas-phase fluorination of PFENin the layer of metal copper and nickel melted calcium fluoride and О±-AI2O3, modified by nickel oxide are listed in Table 4. Here the results on fluorination of PFENin an empty reactor without catalyst are listed for you to compare.
The nonselective, explosive behavior is a specific feature of direct gas-phase fluorination of PFEN in an empty reactor as wells in the layer of metal copper, nickel and melted calcium fluoride at temperature over 440 K according to the experiments; at that tetrafluoromethane and carbon-black are the main products of reactions.
We have obtained nontrivial results while studying gas-phase interaction of PFEN and fluorine at temperatures bellows 420 K – stable radical – 3-perfluoroisopropyl-2-perfluoromethylperfluoropentyl-3 (PFYL) is the main product of reaction in an empty reactor as well as in the layer of metal copper, nickel and melted calcium fluoride (Table 4, # 1-9); we have isolated and characterized it using the method of Electron paramagnetic resonance (EPR). The yield of PFYLat gas-phase fluorination at gas-phase interaction of PFEN and fluorine reaches 85% (per reacted PFEN).
This stable perfluoroalkyl radical was first obtained and characterized using EMR method by the authors /163, 164/ when studying liquid-phase direct fluorination of PFEN; liquid-phase selective fluorination of hexafluoropropylene oligomers is also described in /165,166/. We shall also note, formation of stable perfluoroalkyl radicals at radiolysis and photolysis of perfluoroolefines, particularly, hexafluoropropylene oligomers, is described in /167-173/.
As the studies have proved the extreme stability of PFYL is its characteristic feature – thus, particularly, it is stable during keeping outdoors (6 months). PFYL is stable even in the atmosphere of fluorine up to 420 K (contact period – 20sec) at both their homogenious mixing and contacting in the layer of metal copper, nickel and melted calcium fluoride; when putting the temperature over 440 K the interaction of ПФ�Л and F2shows explosive behavior forming tetrafluoromethane and carbon-black.
The contacting of PFYLwith fluorine in the layer of NiF2/α-Al2O3at 400 K leads to formation of equimolar quantity of 3-perfluoroisopropyl-2-perfluoromethylperfluoropentane (PFAN), and at 520 K and above – to formation of 3-perfluoroethyl-2-trifluoromethylperfluoropentane (PFAO) and tetrafluoromethane.
PFYL is totally decomposing in gas-phase at 470 K in 30 sec forming 3-perfluoroethyl-4-perfluoromethylperfluoropentene-2 (PFEO) and tetrafluormethane; under these conditions while contacting fluorinated О±-Al2O3 PFAOand PFEOare formed along with tetrafluoromethane, as well as PFEN and PFAN:

PFYL also reacts to potassium iodide (aqueous solution, 370 K, 1 hour) isolating iodine and forming PFEN and PFEO:
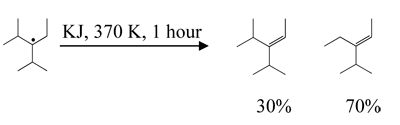
Chemical properties of PFYL are summarized in Table 5.
The principal change of character of direct gas-phase fluorination of PFEN, which is a sharp increase of its rate and total conversion of selectivity was discovered by us when #ing nickel fluoride as a catalyst applied α-Al2O3. Thus, the rate of conversion of PFEN-1 and fluorine at fluorination in an empty reactor, in a layer of metal copper, nickel and melted calcium fluoride (403 K) is 2-5 and 4-13% at a catalyst NiF2/α-Al2O3under the same conditions – more than 99 and 88% respectively (Table 4, # 1, 3, 5, 7 and 12).
Thed fluorination in the layer of nickel fluoride applied on α-Al2O3 at 393-493 K is accompanied by quantitive formation of PFAN – the product of selective attachment of fluorine along the double bond of PFEN (Table 4, # 10-12); PFAN doesn’t virtually form under these conditions at known catalysts such as metal copper and nickel.
Table 5. Some Chemical Properties of PFYL
|
# |
Reaction’s Carrying Out Conditions |
Main Reaction Products |
|
1 |
Air; 298 K, 6 months |
Doesn’t react (stable) |
|
2 |
Fluorine, below 420 K, 20 sec |
Doesn’t react |
|
3 |
Fluorine, Cu, Ni (metal.) CaF2, <420 K |
Doesn’t react |
|
4 |
Fluorine, over 440 K |
CF4, carbon-black |
|
5 |
Fluorine, Cu, Ni (metal.) CaF2, over 440 K |
CF4, carbon-black |
|
6 |
Fluorine, NiF2/О±-Al2O3 , 400 K, 20 sec |
|
|
7 |
Fluorine, NiF2/О±-Al2O3 , 520 K, 20 sec |
|
|
8 |
470 K, 30 sec |
|
|
9 |
О±-AI2O3 (fluorinated), 470 K, 30 sec |
|
|
10 |
KJ, 370 K, 1 hour |
|
It is a rather characteristic feature that the reactivity of PFEN-1, as it follows from the listed data is significantly lower than one of PFEN-2, which is caused apparently by steric particularities of configuration of their molecules.The differences of reactivity of isomers of perfluorononen are clearly illustrated by the results obtained at gas-phase fluorination of their mixture at catalysts of NiF2/О±-Al2O3in the insufficient amount of fluorine (Table 6).
Table 6. Fluorination of Mixture of PFEN-1and PFEN-21 at Catalyst of NiF2/α-Al2O3 (temperature – 403 K, volume rate of input 140 hour-1, mole ratio nitrogen : PFEN = 2,6)
|
## |
Mole Rate Fluorine: PFEN |
Conversion level, % |
Yield
|
||
|
|
|
F2 |
|
||
|
1 |
0,25 |
55,7 |
6,2 |
>99,0 |
98,8 |
|
2 |
0,50 |
80,6 |
28,4 |
>99,0 |
98,4 |
|
3 |
0,80 |
92,8 |
70,1 |
>99,0 |
98,0 |
|
4 |
1,20 |
>99,9 |
>99,9 |
88,4 |
97,4 |
1Composition of initial mixture, mass.% ![]() -33,2;
-33,2; ![]() -66,2.
-66,2.
В
Fluorination of PFEN in the layer of NiF2/О±-Al2O3at high temperatures (420 K and above) leads to increase of formation of PFAO (Table 4, # 13), apparently, due to elimination of trifluoromethyl group at interimly forming PFYL and following selective fluorination of intermediate PFEO with obtaining PFAO:
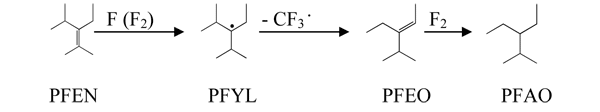
Here we must remind, that fluorination of PFEN at known catalysts – metal copper and nickel under these conditions (temperature above 440 K) passes nonselctively with formation mainly of tetrafluormethane and carbon-black.
The obtained results fully conform to main positions of offered model of selective catalytic fluorination. Indeed, the localization of fluorination on the surface of catalyst NiF2/О±-Al2O3 led to abrupt drop in destructive fluorination and increase of process selectivity; the absence of deep destruction during fluorination under severe conditions (440 k and above) so much typical for fluorination in the layer of metal nozzles and empty reactor, is caused, apprently, by in-time removing of excessive energy of interimly forming exited adducts along the bonds with crystal lattice/matrix of hetherogenic contact.
The presence of atomic fluorine on a rather developed surface of NiF2/α-Al2O3explains both sharp increase of the process rate compare to the known catalysts (this very thing leads to the localization of fluorination on the surface)and specific features of fluorination selectivity: interaction of PFEN and molecular fluorine (in the layer of inert nozzle in full rector) sterucally is hardened and passes slowly and stops at the stage of PFYL formation. Specific surface of waste/used catalyst NiF2/α-Al2O3is 2,5 – 3,0 m2/ml and approximately exceeds by a factor of hundred the one of metal catalyst. Further fluorination of PFYL is only possible when atomic fluorine is participating (due to its smaller geometrical sizes), that is confirmed by particularities of fluorine interaction with both PFYL, and PFEN in the layer of NiF2/α-Al2O3.
The nature of catalitic action of NiF2/О±-Al2O3 at direct gas-phase fluorination of PFEN is caused thus by the activation of fluorine adsorbed and, as a consequence, by localization of fluorine acts on the surface; this principally differs from the function of metal nozzles boiling down to removal (dispersion) of thermal energy isolating at fluorination.
From the practical point of view high rate and selectivity of direct gas-phase fluorination of PFEN in the layer of allows considering this process as a base for effective PFAN obtaining technology.
2.3.2 Perfluorohexenes’ Fluorination
Selective fluorination of rather available perfluorohexenes – cis- and trans-isomers of 2-perfluoromethylperfluoropentene-2 obtained using anionic dimerization of hexafluoropropylene is one of the possible ways to synthesize 2-perfluoromethyl perfluoropentane – an effective dielectric liquid.Information about direct gas-phase fluorination of those polyfluorinated olefins based on catalyst NiF2/α-Al2O3are listed in Table 7; here the results obtained using known metal nickel catalyst are also given for comparison.
Table 7. Direct Fluorination of Perfluorohexenes (temperature – 373 K, volume rate of input – 220 hour-1, mole ratio of subsrate:fluorine:nitrogen as 1:1,2:2,5).
|
# |
Catalyst |
Reaction Products’ Composition, volume % |
||||||
|
CF4 |
C2F6 |
C3F8 |
C4F10 |
C5F12 |
C6F14 |
C6F12 |
||
|
1 |
Ni,Metal |
15,4 |
6,1 |
6,4 |
2,1 |
3,4 |
36,8 |
29,8 |
|
2 |
NiF2/О±-Al2O3 |
6,3 |
3,2 |
4,1 |
1,9 |
1,6 |
82,9 |
<0,1 |
Looking at data given here it is seen, that direct gas-phase fluorination of perfluorohexenes at catalyst NiF2/О±-Al2O3 goes rather selectively; the yield of perfluorohexane at metal nickel catalyst is significantly lower.
In case of fluorination of perfluorohexenes there are no steric difficulties, that’s why data for fluorination at #ed heterogeneous contacts of principally different nature of action differ only in quantitive terms.
WE shall also note, that listed results allow consicdering direct catalytic fluorination as a promising method to obtain 2-perfluoromethylperfluoropentane based on isomers of 2-perfluoromethylperfluoropentene-2.
2.3.3 Perfluorobutenes’ Fluorination
Data on direct gas-phase fluorination of perfluorobutenes in the mixture with octafluorocyclobutane and perfluoromethylperfluorocyclobutane over melted calcium fluoride, metal copper, as well as nickel fluoride applied on О±-Al2O3, is listed in Table 8; here the results on fluorination in an empty reactor without catalyst are listed for comparison.
Tetrafluoromethane and carbon-black are the main products of reaction without catalyst; at that the process is promt to explosion even at 300 K. The selective attachment of fluorine along double bond doesn’t occur; octafluorocyclobutane and perfluoromethylperfluorocyclobutane are subject to destructive fluorination along with perfluorobutenes. The last fact mentioned conforms to data /174/, where it is shown that at 550 K and above the fluorine reacts to octafluorocyclobutane under homogeneous conditions formingenergetically exited intermediate radical n-C4F9•, which main product of further transformations is tetrafluoromethane.
Using nozzle made of melted calcium fluoride or metal copper leads to some increase of fluorination selectivity
(Experiments 3-6, Table 8) – the yield of perfluorobutane reaches 50%. At the same time in this case too the process is characterized by deep destruction of raw materials forming tetrafluoromethane.
The principal increase of fluorination selectivity is observed when carrying out the process in the layer of NiF2/α-AI2O3 – the yield of perfluorobutane increases up to 96-99% at perfluorobutenes degree of conversion exceeding 99%; octafluorocyclobutane and perfluoromethylperfluorocyclobutane virtually do not react during that.
2.3.4 Hexafluoropropylene Fluorination
To estimate the effect of fluorine’s activation by catalysts’s surface onto selectivity of its attachment along the double bond gas-phase fluorination of hexafluoropropylene had been studied as a model reaction as well:
C3F6 + F2 = C3F8 – 635 kJ/mole
Nickel fluoride applied onto α-AI2O3 – contact asserting the activation of fluorine on the surface had been #ed as a catalyst and for comparison unmodified α-AI2O3, melted calcium fluoride and known metal catalysts – copper and nickel. The part of experiments had been conducted in an empty reactor. The information can be found in Table 9.
On can see, that interaction of fluorine and hexafluoropropylene in an empty reactor (even when it is diluted with nitrogen) passes in a non-selective way – tetrafluoromethane is the main product of reaction. The filling of reactor with nozzle/packing results in specific (depending on the nozzle’s nature) increase of the input of selective fluorination. When using nickel fluoride applied onto α-AI2O3, (# 11,12, Table 9) the hexafluoropropylene fluorination passes in a most selective way; only in that case octafluoropropane is the main product even when using undiluted fluorine.Apparently, that result is a consequence of the increase of the input of selective heterogeneous fluorination at activation of fluorine at dispersed nickel difluoride.
The association of intermediate radicals forming significant quantities of limear and branhed perfluorohexanes is a characteristic feature of hexafluoropropylene’s direct fluorination.
Table 9. Hexafluoropropylene Direct Fluorination .
2.3.5 Tetrafluoroethylene and Trifluorochloroethylene Fluorination
In Tables 10 and 11 comparative data on composition of products of direct fluorination of tetrafluoroehylene and trifluorochloroethylene at catalyst of NiF2/α-Al2O3 and in the layer of dense nozzle/packing made of thin copper.During fluorination of tetrafluoroethylene and trifluoroethylene the volume rate of input was 220 hour-1, mole ratio subsrate:fluorine: nitrogen = 1:1,1:2,5; temperature of reactor’s exterior wall was 313 K (at heating off).
Table 10. Direct Fluorination of tetrafluoroethylene.
|
# |
Catalyst |
Fluorination Product Composition, volume% |
|||||
|
CF4 |
C2F6 |
C3F8 |
C4F10 |
C5F12 |
Others |
||
|
1 |
NiF2/О±-Al2O3 |
24,8 |
34,7 |
12,8 |
12,4 |
4,2 |
11,1 |
|
2 |
Cu, metal |
37,7 |
22,4 |
10,7 |
11,8 |
4,0 |
13,4 |
One can see from the listed data, that direct fluorination of tetrafluoroethylene and trifluorochloroethylene passes nonselectivelyforming significant quantities of dimerization and destruction products; the yield of selective fluorination products – hexafluoroethane and pentafluorochloroethane at NiF2/α-Al2O3 , although, is significantly higher, than when using metal nozzle/ packing.
Table 11. Trichloroethylene Direct Fluorination.
|
# |
Catalyst |
Fluorination Products Composition, volume. % |
|||||||
|
CF4 |
C2F6 |
CF3Cl |
C3F8 |
C2F5Cl |
C4F10 |
C4F8Cl2 |
Others |
||
|
1 |
NiF2/О±-Al2O3 |
10,6 |
4,2 |
9,6 |
5,7 |
33,6 |
2,5 |
17,3 |
16,5 |
|
2 |
Cu; metal |
18,5 |
4,1 |
13,4 |
7,1 |
20,8 |
2,8 |
16,8 |
16,5 |
While data on direct gas-phase fluorination of hexafluoropropylene and, particularly, of tetrafluoroethylene
and trifluorochloroethylene testiifies the increase of input of selective fluorination when using
developed catalyst NiF2/О±-Al2O3 (compare to known metal contacts)
it clearly illustrates the limitations of that method.
Low selectivity of fluorination of “small” fluorolefines can be caused by significant heating up of reaction gases during fluorination (due to their low thermal capacity), when thermal decomposition contributes mainly to the destruction, and not the decomposition of energeticall y exited molecules ( as in case with perfluorononenes and perfluorobutenes).
Due to that the studying of direct catalytic fluorination of fluorolefines diluted with inert, saturated fluororganic compounds was of our interest; such process’s carrying out would have allowed decreasing the heating up of reaction gases in exothermal fluorination and thus lowering the input of thermal destruction.
2.3.6 Two-stage Olefines’ Fluorination. Olefines Removal from Fluorochlorocarbons
The heating up of reaction gases in direct fluorination can be decreased by carrying out preliminary “soft” fluorination of olefins, for example, using cobalt trifluoride.Thereupon we had studied two-phase fluorination of tetrafluoroethylene, acethylene, hexafluoropropylene and trifluorochloroethylene /175/. We shall note, that the authors /176-179/ had studied in details the fluorination of acetylene and hexafluoropropylene using cobalt trifluoride, where it is shown, that the yield of hexafluoroethane at fluorination of acetylene is small because of forming partial fluorination products in significant quantities – pentafluoroethane and 1,1,2,2-tetrafluoroethen – hydrogen containing adducts, which further fluorination using cobalt trifluoride goes rather slowly. At the same time the obtaining of hexafluoroethane a precious working body for plazmo- chemical processing of elements of electronic systems, out of acetylene would allow carrying out the synthesis with minimum spending of luorine without using intermediate chloroorganic products.The obtaining technology for tetrafluoroethylene, from which hexafluoroethane can be obtained, includes the use of chloroform, which production is rather labour intensive and is characterized by significant chlorine spending.
In the present part of the work the data on direct catalytic fluorination of the following products is listed as well:
- tetrachloroethylene and fluorotrichloroethylene in the mixture with fluorochlorocarbons of ethane row (chladones – 111, - 112 and 113); such mixtures are formed at fluorination of tetrachloroethylene by urane hexafluoride – the presence of haloidolefines in them complicates the following utilization of chladones;
- unsaturated and hydrogen containing addmixtures in octafluorocyclobutane for the purpose of purifying that perspective solvent for co-polymerization of tetrafluoroethylene and perfluoropropylperfluorovinyl ether.
Obtaining of Hexafluoroethane out of Acetylene
Data on acetylene fluorination by cobalt trifluoride can be found in Table 12.
Table 12. Fluorination of acetylene using cobalt trifluoride (input rate of acetylene – 3 l/h, amount of cobalt trifluoride – 250 g active fluorine contain 13, 2 mass.%).
|
# |
T, K |
Time from experiment start, hour, |
Reaction Gases Composition, volume % |
|||||||
|
CF4 |
C2F6 |
C2H2 |
C2F5H |
CH2F-CF3 |
CHF2- CHF2 |
C4F10 |
Others |
|||
|
1 |
473 |
1 |
<0,1 |
0,5 |
1,7 |
25,3 |
7,2 |
62,3 |
<0,1 |
3,0 |
|
2 |
473 |
2 |
<0,1 |
0,3 |
4,8 |
21,8 |
8,8 |
60,1 |
<0,1 |
4,2 |
|
3 |
573 |
1 |
<0,1 |
17,2 |
0,3 |
47,0 |
1,8 |
32,3 |
<0,1 |
1,4 |
|
4 |
573 |
2 |
<0,1 |
6,0 |
4,3 |
46,1 |
3,8 |
38,3 |
<0,1 |
1,5 |
|
5 |
623 |
1 |
0,3 |
72,9 |
0,1 |
22,2 |
0,5 |
1,1 |
1,6 |
0,7 |
|
6 |
623 |
2 |
0,3 |
51,5 |
2,2 |
40,9 |
0,9 |
1,8 |
1,2 |
1,5 |
From the data above we can see, that fluorination within the temperature range of 470 – 625 K goes virtually without raw materials destruction – main products of reaction are 1,1,2,2-tetrafluoroethane, pentafluoroethane (within temperature range of 470-570 K) and hexafluoroethane (at 620 K and above). It is typical, that along with the product of selective fluorine attachment – I,I,2,2-tetrafluoroethane significant amounts of 1,1,1,2-tetrafluoroethane are formed; analogous re-distribution of hydrogen in the products of ethylene fluorination using cobalt trifluoride had been noticed by the authors /180/.
Decreasing of content of active fluorine in cobalt fluoride observed in the process leads to cutting of fluorination (Table 12, experiments 2,4,6).
By the listed data we can see, that even at 623 K the content of hexafluoroethane in reaction gases doesn’t exceed 75 volume %; further increase of temperature leads to intensive forming of carbon and tetrafluoromethane. We shall note, that these results conform the data on fluorination of acetylene by cobalt trifluoride listed in /177/.
In Table 13 we can find the data on direct fluorination of acetylene and cobalt trifluoride interaction products.
Table 13. Direct Fluorination of Acetylene And Cobalt Trifluoride Interaction Products At Catalyst NiF2/α-Al2O3 (reactor volume – 50 ml, input rate: organic products in experimentss 1-3 – 2 l/h, in # 4 – 1 l/h, fluorine – 1,5l/h).
| # |
Temperature, K |
Reaction Gases Composition, volume % |
||||||||||
|
CF4 |
C2F6 |
C2H2 |
C2F5H |
CH2F- CF3 |
CHF2- CHF2 |
C4F10 |
Others |
|||||
|
Initial Products' Composition |
0,3 |
72,9 |
<0,1 |
22,5 |
0,5 |
1,1 |
1,6 |
В 0,7 |
||||
|
1 |
473 |
0,3 |
97,0 |
<0,1 |
0,4 |
<0,1 |
0,2 |
1,5 |
В 0,6 |
|||
|
2 |
523 |
0,3 |
97,4 |
<0,1 |
<0,1 |
<0,1 |
<0,1 |
1,6 |
В 0,7 |
|||
|
3 |
573 |
0,3 |
97,3 |
<0,1 |
<0,1 |
<0,1 |
<0,1 |
1,6 |
В 0,8 |
|||
|
Initial Products' Composition |
<0,1 |
6,0 |
4,3 |
46,1 |
3,8 |
38,3 |
<0,1 |
В 1,5 |
||||
|
4 |
523 |
25,1 |
72,0 |
<0,1 |
1,4 |
<0,1 |
<0,1 |
<0,1 |
В 1,5 |
|||
By the data above we can see, that direct fluorination of acetylene and cobalt trifluoride interaction products at catalyst NiF2/α-Al2O3 is accompanied by selective fluorination of pentafluorochloroethane and allows obtaining of perfluorinated products containing 97,0 – 97,4 volume % of hexafluoroethane (#s 1 – 3, Table 13); fluorination of mixtures containing significant amounts of tetrafluoroethane and unreacted acetylene is accompanied by forming of tetrafluoromethane (# 4, Table 13).
We shall note, that tetrafluoroethane, pentafluoroethane and hexafluoroethane can be obtained of virtually quantitive yield as shown in /175/ by such two-phase fluorination of tetrafluoroethylene, trifluorochloroethylene and hexafluoropropylene respectively (at first by cobalt trifluoride, and then by fluorine at NiF2/О±-Al2O3); at that, the content of haloidolefines in the target products was lower than the level of sensitivity of their discovery (0,01 volume %).
Data on direct fluorination of tetrachloroethylene and fluorotrichloroethylene in the mixture with fluorochlorocarbons of ethane row are listed in Table 14.
Table 14. Direct Fluorination of Tetrachloroethylene and Fluorotrichloroethylene in the Mixture With Chladones Of Ethane Row At Catalyst NiF2/О±-Al2O3.*
|
# |
T, K |
Organic Products Composition, mass % |
|||||
|
C2F4Cl2 |
C2F3Cl3 |
C2F2 Cl4 |
C2Cl4 |
C2 Cl3F |
Others |
||
|
Initial Products Composition |
0,3 |
91,8 |
0,3 |
4,8 |
2,6 |
0,2 |
|
|
1 |
333 |
0,3 |
94,3 |
4,6 |
0,4 |
0,1 |
0,3 |
|
2 |
393 |
0,4 |
95,2 |
4,2 |
<0,1 |
<0,1 |
0,3 |
|
3 |
573 |
3,3 |
92,8 |
3,5 |
<0,1 |
<0,1 |
0,4 |
|
Initial Products Composition |
<0,1 |
24,3 |
60,2 |
12,2 |
0,2 |
0,2 |
|
|
4 |
453 |
0,1 |
27,9 |
71,8 |
<0,1 |
<0,1 |
0,2 |
|
5 |
473 |
0,3 |
28,2 |
71,4 |
<0,1 |
<0,1 |
0,4 |
|
6 |
523 |
0,8 |
31,4 |
67,5 |
<0,1 |
<0,1 |
0,3 |
*Experiments 1-3 were carried out in the reactor of 50 ml volume, input rate of organic products – 100 g/hour, fluorine – 2 g/hour. Experiments 4-6 were carried out in the pilot reactor volume 6,2 l, input rate of organic products – 10 kg/hour, fluorine – 0,3 kg/hour, nitrogen – 4m3/hour.
From the data above we can see, that fluorination of tetrachloroethylene and fluorotrichloroethane at catalyst NiF2/α-Al2O3 goes in a selective way forming 1,2-difluorotetrachloroethane and 1,1,2-trifluorotrichloroethane respectively and within the temperature range 390-570 K and it is characterized by total purification of unwanted haloidolefiens; at that the fluorination of saturated fluorochlorocarbons doesn’t occur.
We shall note, that purification of ethane row chladones from tetrachloroethylene and fluorotrichloroethylene using the method of direct catalytic fluorination had been successfully #ed at a commercial scale (reactor of 60l volume).
Data on purification of octafluorocyclobutane from unsaturated and hydrogen containing addmixtures by direct catalytic fluorination are listed in Table 15.
Table 15. Octafluorocyclobutane’s Purification From Unsaturated And Hydrogen Containing Addmixtures by Direct Fluorination At Catalyst (Volume Velocity of Input - 60 hour-1, mole ratio octafluorocyclobutane:fluorine= 20).
|
Addmixtures’ Titles |
Addmixtures Content in Octafluorocyclobutane, volume % |
Addmixtures Content volume (%) in octafluorocyclobutane, purified at, K |
|
|
423 |
473 |
||
|
hexafluoropropylene |
0,24 |
9x10-4 |
<10-4 |
|
perfluorobutyne-2 |
6x10-4 |
<10-4 |
<10-4 |
|
2,2-dihydroperfluoropropane |
2x10-4 |
10-4 |
<10-4 |
|
2-hydroperfluoropropylene |
9x10-4 |
4x10-4 |
<10-4 |
|
trans-perfluorobutene -2 |
9,7x10-2 |
<5x10-3 |
<5x10-3 |
|
cis-perfluorobutene-2 |
2,1x10-2 |
<5x10-3 |
<5x10-3 |
|
perfluorocyclobutene |
1,2x10-2 |
<10-4 |
<10-4 |
From the data above we can see, that method of direct catalytic fluorination allows carrying out fine purification of octafluorocyclobutane from unsaturated and hydrogen containing addmixtures. As experiments proved /181/, purified octafluorocyclobutane is an effective solvent for carrying out the co-polymerization of perfluoropropylperfluorvinyl ether and tetrafluoroethylene obtaining fluoroplast-50.
Thus listed results show, that developed catalyst NiF2/α-Al2O3 allows carrying out direct selective fluorination of “small” fluorolefines at diluting them with inert saturated fluororganic compounds.These results open new practical perspectives for obtaining pure fluorocarbons based on corresponding olefins: their synthesis can be carried out in two stages and includes preliminary fluorination at cobalt trifluoride and following direct catalytic fluorination.
The developed method can be used for fine purification of fluorocarbons from underfluorinated compounds.
2.3.7 Fluorination Of Perfluoropropionylfluoride and Hexafluoropropyleneoxide Lowest Oligomers. Stabilization of Perfluoropolyesters Based on Hexafluoropropylene and Oxygen
The technology of inert perfluoropolyethers – oxygen containing fluorocarboxylic liquids based on hexafluoropropylene and oxygen widely used in special fields of technics includes fluorination of mixture of polymolecular products of low-temperature initiated oxidation containing end fluoranhydride group; as a result an elimination of carbonyldifluoride and stabilization of perfluoropolyethers takes place /182-184/:
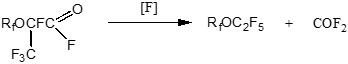
В
Highmolecular perfluoropolyethers are stabilized by fluorine in liquid phase, and low-molecular – in gaseous phase using cobalt rtifluoride; in both cases the fluorination is characterized by high destruction of precious fluorocarbonic raw materials.
Thereupon and for the purpose of establishing the impact of heterogeniuos contact nature onto selectivity and rate of interaction of fluoroanhydrides and fluorine we have studied model reactions of direct fluorination of perfluoropropionylfluoride and individual perfluoropolyethers containing end fluoroanhydride group – lowest oligomers of hexafluoropropylene oxide: 2-perfluoromethyl-3-oxaperfluorohexanoilfluoride (hexafluoropropylene oxide dimer), 2,5-perfluorodimethyl-3,6-dioxaperfluorononoylfluoride (hexafluoropropylene oxide trimer) and 2,5,8-perfluorotrimethyl-3,6,9-trioxaperfluorononoylfluoride (hexafluoropropylene oxide trimer) and 2,5,8-perfluorotrimethyl-3,6,9-trioxaperflurododecanoylfluoride (hexafluoropropylene oxide tetramer) – low-molecular analogues of perfluoropolyethers based on hexafluoropropylene and oxygen.
Here we shall note, that the direct gas-phase fluorination of perfluoropropionylfluoride and oligomers of hexafluoropropylene oxide in an empty reactor, without catalyst is explosive and uncontrolled – tetrafluoromethane, carbonyldifluoride and carbon-black are the main products of reaction. Information on direct gas-phase fluorination of perfluoropropionylfluoride and lowest oligomers of hexafluoropropylene oxide over metal nickel, α-AI2O3 and NiF2/α-Al2O3 catalyst can be found in Table 16,17 and at Picture 2.
Table 16. Direct Catalytic Fluorination of Perfluoroproppionylfluoride (volume rate of input is 95 hour-1, mole ratio fluorine:substrate=1,2).
|
# |
Catalyst |
Temperature, K |
Conversion level PF1,% |
Neutralized Reaction Gases Composition2, Volume % |
||
|
CF4 |
C2F6 |
Others |
||||
|
1 |
Ni, metal. |
473 |
14 |
45,6 |
52,3 |
2,1 |
|
2 |
О±-AI2O3 |
473 |
22 |
39,4 |
57,1 |
3,6 |
|
3 |
NiF2/О±-Al2O3 |
373 |
34 |
35,3 |
60,8 |
3,9 |
|
4 |
Same |
473 |
80 |
20,6 |
79,0 |
0,4 |
1The Conversion level of perfluoropropionylfluoride at 373 K at О±-Al2O3and metal nickel is lower than 1%.
2Composition of neutralized reaction gases is given without taking carbon dioxide into account.
By the information listed in Table 16 one can see, that rate and selectivity of fluorinaion of perfluoropropionylfluoride significantly depends on the nature of applied catalyst; NiF2/О±-Al2O3catalyst is most active.
Hexafluoroethane and tetrafluoromethane are the main reaction products; it is typical, that when using less active catalysts of AI2O3 and metal nickel under comparable conditions the α- relative yield of product of deep destructive fluorination – tetarfluoromethane is significantly higher than at NiF2/α-Al2O3.
One can see from the data listed in Table 17 and on Picture 2, that the nature of heterogeneous contact impacts significantly the fluorination rate of lowest oligomers of hexafluoropropylene oxide.
Thus, the Conversion level of hexafluoropropylene oxide trimer at О±-AI2O3, metallic nickel and NiF2/О±-Al2O3 at 473 K is 11, 35 and 90 %, respectively (#s 10,13,16, Table 17). Practically quantitive yield of 5,8-perfluorodimethyl-3,6,9-trioxaperfluorododecane at fluorination of hexafluorop[ropylene oxide tetramer at catalyst NiF2/О±-Al2O3draws our attention (# 17, Table. 17).
Catalytic gas-phase fluorination of lowest oligomers of hexafluoropropylene oxide can contain the formation of unstable perfluoroalkylhypofluorites, which further destruction in accordance with /185/ passes with elimination of carbonyldifluoride and forming of stable fluoroethers with end perfluoroethoxyl group. This conjection conforms to the pointed earlier higher activity of NiF2/О±-Al2O3 catalyst during the synthesis of trifluoromethylhypofluorite out of carbon oxide or carbonylfluoride.
From the listed in Table 17 data one can see, that reactivity of lowest oligomers of hexafluoropropylene oxide is not equal – thus, at 473 K the rate of conversion of dimer, trimer and tetramer at NiF2/α-Al2O3 catalyst amounted to 52, 90 and 98% respectively. The increasing of reactivity of fluoroanhydrides along with (simalteneously) their molecular mass growth is rather important from the practical point of view and it indicates the higher reactivity of relatively high-molecular perfluoropolyethers forming during low temperature initiated oxidation of hexafluoropropylene. However, gas-phase fluorination of high-molecular perfluoropolyethers is possible only at low pressure or dilution with inert gas.
Table
17. Direct
Catalytic Fluorination of Lowest Hexafluoropropyleneoxide Oligomers
(volume rate of input - 95 hour-1,
mole ratio fluorine:substrate = 1,2).
|
# |
Catalyst |
Reaction Temperature, K |
Conversion level of Organic Substrate % |
Main Product Yield ,1% |
|
1. Fluorination of Dimer of HFPO |
||||
|
1 |
Ni, metallic |
423 |
9 |
97 |
|
2 |
Same |
473 |
15 |
91 |
|
3 |
О±-AI2O3 |
423 |
3 |
98 |
|
4 |
Same |
473 |
7 |
98 |
|
5 |
NiF2/О±-Al2O3 |
393 |
14 |
99 |
|
6 |
Same |
423 |
28 |
99 |
|
7 |
- В«- |
473 |
52 |
98 |
|
2. Fluorination of Trimer of HFPO |
||||
|
8 |
Ni, metallic |
393 |
6 |
98 |
|
9 |
Same |
423 |
16 |
98 |
|
10 |
-В«- |
473 |
35 |
92 |
|
11 |
О±-AI2O3 |
393 |
2 |
99 |
|
12 |
Same |
423 |
4 |
99 |
|
13 |
-В«- |
473 |
11 |
98 |
|
14 |
NiF2/О±-Al2O3 |
393 |
22 |
99 |
|
15 |
Same |
423 |
53 |
99 |
|
16 |
-В«- |
473 |
90 |
98 |
|
3. Fluorination of Tetramer of HFPO |
||||
|
17 |
NiF2/О±-Al2O3 |
473 |
98 |
96 |
13-oxaperfluorohexane, 5-perfluoromethyl-3,6-dioxaperfluorononane and 5,8-perfluorodimethyl-3,6,9-trioxaperfluorododecane are the main products of selective fluorination of dimer, trimer and tetramer of HFPO respectively.
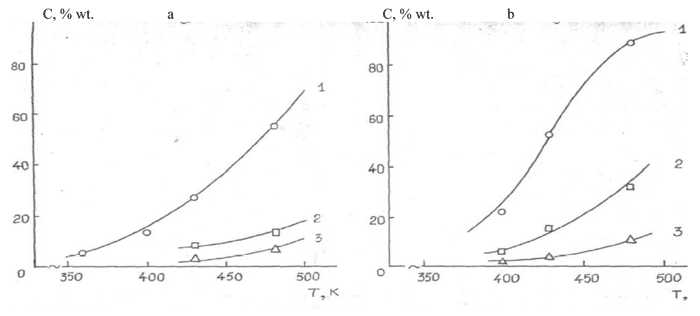
Picture 2.Temperature Dependancies of concentrations of 3-oxaperfluorohexane (a) and 5-perfluoromethyl-3,6-oxaperfluorononane (b) in liquid products of direct gas-phase fluorination of hexafluoropropyleneoxide’s dimer and trimer at catalysts: 1 - NiF2/α-Al2O3; 2 – metal nickel and 3 - α-AI2O3 respectively.
The effective catalyst NiF2/О±-Al2O3 for fluorination of lowest oligomers of hexafluoropropylene oxide had been #ed for gas-phase stabilization of light fraction of perfluoropolyethers based on hexafluoropropylene and oxygen (boiling out temperature 313-343 K at 133 Pa). Nowdays stabilizing technology of this fraction is based, as it has been mentioned, on fluorination of cobalt trifluoride and is characterized by low productivity, high material and energy consumption. Comparative data on fluorination of perfluoropolyethers using fluorine at NiF2/О±-Al2O3 catalyst and using cobalt trifluoride are listed in Table 18.
Table 18. Stabilization of Light Fraction of Perfluoropolyethers Based on Hexafluoropropylene and Oxygen.
|
# |
Temperature, K |
Raw Materials Loss at Fluorination, mass.% |
Residual Concentration of detached Ion Fluorine1, mass. % |
Degree of Fluorination % |
|
1. Direct Fluorination of Catalyst NiF2/О±-Al2O32 |
||||
|
1 |
573 |
7,1 |
1,0 x 10-1 |
91,3 |
|
2 |
623 |
8,5 |
8,2 x 10-3 |
99,3 |
|
3 |
643 |
9,7 |
4,3 10-3 |
99,6 |
|
4 |
673 |
10,6 |
8,0 x 10-4 |
>99,9 |
|
2. Fluorination using Cobalt Trifluoride3 |
||||
|
5 |
573 |
16,3 |
2,5 x 10-1 |
78,3 |
|
6 |
623 |
18,7 |
1,4 x 10-1 |
87,8 |
|
7 |
673 |
20,8 |
5,5 x 10-2 |
95,2 |
1The content of fluoroanhydride groups was estimated by concentration of ion-fluoride detached during prolonged thermal water-alkali hydrolysis; its content in initial ethers amounted to 1,15 mass. %.
2Catalyst amount– 50ml, input rate of ethers – 50 g/hour, fluorine – 4 g/hour, fluorine Conversion level at 573, 623, 643 and 673 K amounted to 44, 51, 58 and 65 %, respectively.
3Cobalt trifluoride amount – 250 g, content of active fluorine – 13,4 mass.%, ethers’ input rate – 50 g/hour.
Listed data clearly illustrate the advantages of catalytic method, which lie in high rate and selectivity of fluorination. Raw materials loss at catalytic fluorination under rather severe conditions (673 K) amounted only to 10,6 mass.% (# # 4, Table 18), which is really lower, than at using cobalt trifluoride under analogous conditions (20,8 mas..% - # 7, Table 18).
The difference in fluorination rates can be estimated by residual content of unreacted fluoroanhydrides (they were determined using concentration of detached ion-fluorine in fluorinated ethers) – their number in products of direct catalytic fluorination is more than 50 times lower than at cobalt trifluoride using (# 4,7 Table 18).
to be continued
References
2. Tedder D.M. Ftorirovanie organicheskih soedinenij ehlementarnym ftorom. V kn."Uspekhi khimii ftora", t. I-II -M.-L. : Khimiya, 1964, p. 380-423.
3. Orkin V.L., Chaikin A.M. Opredelenie konstant skorosti obrazovaniya atomov v reakciyah molekulyarnogo ftora s okis'yu azota, ehtilenom i tetraftorehtilenom. Kinetika i kataliz, 1982, t. 23, v. Z, p. 529-533.
4. Anson P.O., Fredricks P.S., Tedder J.M. Free-radical substitutionin aliphatic compounds. Part 1. Halogenation of n-butahe and isobutane in the gas phase. J.Chem.Soc, 1959, March, p.918-922.
5. Fredricks P.S., Tedder. J.M. Free-radical substitution in aliphatic compounds. Part 11. Halogenation of the n-butylhalides. -J.Chem.Soc, 1960, Jan., p. 144-150.
6. Purington S.T., Kagen B.S., Patric T.B. The application of elemental fluorine in organic synthesis. Chem.Rev., 1986, № 86, p.997-1018.
7. Aikman R.E., Lagow E.J. Syhthesis of tetra-cis-(perfluorocyclohexyl)-methane and bis-(perfluorocyclohexyl)-methane by direct fluorination. -J.Org.Chem., 1982, v.47, № 14, p.2789-2790.
8. Eremenko L.T., Oreshko G.V. Ftorirovanie ehlementarnym ftorom labil'nyh polinitrosoedinenij – adduktov reakcii Mihaehlya. -Izv. AN SSSR, Ser. khim, 1969, № 2, p.479.
9. Eremenko L.T., Nacibullin F.Ya., Borovinskaya J.P., Karpova N.D. Sintez perftornitroehtanov. -Izv. AN SSSR. Ser. khim., 1968, № 2, p.429-430.
10. Patent US 4523039. Sposob polucheniya perforirovannyh prostyh ehfirov. /Lagov R.J., Gerhart J.E. -Publ. 11.06.85, RZh Khimiya, 1986, 7N36.
11. Patent US 3242218. Sposob polucheniya ftoruglerodnyh poliehfirov. /Miller V.T. -Publ. 22.03.66, RZh Khimiya, 1967, 12C232.
12. Des Martean D.D, Fluoroperoxytrifluoromethane CF300F. Preparation from trifluoromethyl hydroperoxide and fluorine in the presence of cesium fluoride. -Inorg Chem., 1972, v.11, № 1, p.193-195.
13. Merritt R.F., Johnson F.A. Direct fluorination. Addition of fluorine to indenes and.acenaphthylenes, -J.Org.Chem., 1966, v.31. № 6, p.1859-1863.
14. Merritt R.F, Johnson F.A. Direct fluorination of steroidal olefines to cis-vicinal difluorides. –J.Am.Chem.Soc., 1966, v.88, № 8, p.1822-1823.
15. Patent US 3487093. Ftorirovannye olefiny. Merrit R.F. –Publ. 30.12.69, RZH Khimiya, 1970, 23N29.
16. Grakauskas V. Direct liquid phase fluorination of halogenated aromatic compounds. -J.Org.Chem., 1969, v.34, № 10, p.2835-39.
17. Brooke G.M., Chambers B.D., Heyes J., Musgrave W.K.R. Direct preparation and some reactions ofсhlorofluorobenzenes. -J.Chem.Soc., 1964, Febr., p.729-735.
18. Merritt R.F, The polar addition of molecular fluorine to acetylenes. –J.Org.Chem., 1967, v.32, № 12, p.4124-4126.
19. Sheppard U., Sharts K. Organicheskaya Khimiya ftora. -
M.: Mir, 1972, p. 52, 89, 112.
20. Miller V., Ehrenfeld J, Felan J., Prober M., Rid Sh, Ftorirovanie polnost'yu galoidirovannyh olefinov. «Khimiya ftora», № 2, -M.: IL, 1950, p. 228-240.
21. Miller W.T. My early days in fluorine chemistry. –J.Fluor.Chem., 1981, v.18, p.305-321.
22. Miller W.T., Stoffer J.O., Fuller G., Currie A.C. The mechanism of fluorination. IV. The effect of temperature and of fluorine concentration on reaction. A new fluorination apparatus. –
J.Am.Chem.Soc., 1964, v.86, № 1, p.51–56.
23. Isikava Nobuo, Kitacumeh Tomoya. Novye metody ftorirovaniya aromaticheskih soedinenij. –Yukki gosehj kachaku kyokajsi., 1976, t.34, № 3, p.173-178, RZh Khimiya, 1976, 23ZH360.
24. Patent JP 55-18695. Sposob ftorirovaniya./ Maruo Kehjiti, Misaki Susumu. –Publ. 21.05.80, RZh Khimiya, 1981, 5N122.
25. Sirip L.A., Lagow R.J. Direct fluorination of 2,2,4,4-tetramethyl pentane. Sterically protected residual protons. –J.Org.Chem., 1977, v.42, № 21, p.3437-3438.
26. Kowanko N., Branthaver J.F., Sugihara J.M. Direct liquid-phase fluorination of petroleus. –Fuel, 1978, v.57, № 12, p.769-775.
27. Patent US 40004996. Ftorirovanie organicheskikh soedinenij./ Kollonic J. –Publ. 25.01.77, RZh Khimiya, 1977, 23N57.
28. Patent GB 1077065. Ftorirovanie nitrosoedinenij./ Grakauskas V., Hemel E.E. –Publ. 26.07.67, RZh Khimiya, 1975, 16N84.
29. Merritt R.F. Direct fluorination of 1,1-diphenylethylene. –J.Org.Chem., 1966, v.31, № 11, p.3871-3873.
30. Merritt R.F. The polar fluorination of propenylbenzene. –J.Am.Chem.Soc., 1967, v.89, № 3, p.609-612.
31. Merritt R.F., Johnson F.A. Low-temperature fluorination of Schiff bases. -J.Org.Chem., 1967, v.32, № 2, p.416-419.
32. Misaki S. Direct fluorination of phenol and cresols. –
J.Fluor.Chem., 1981, v.17, № 2, p.159-171.
33. Maxwell A.F., Detoro F.E., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The jet fluorination of certain aliphatic hydrocarbons as oriented and controlled by operation conditions. -J.Am.Chem.Soc., 1960, v.82, № 22, p.5827-5830.
34. Attaway J.A., Groth R.H., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The fluorination of some amides, nitriles and of methylthiocyanate. -J.Am.Chem.Soc., 1959, v.81, № 14, p.3599-3603.
35. Robson P., Mc Longhlin V.C.R., Hynes J.B., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIV. The jet fluorination of hydrogen cyanide, cyanogen, methylamine and ethylenediamine. Pyrolysis and fluorinolysis of selected products. -J.Am.Chem.Soc., 1961, v.83, № 24, p.5010-5015.
36. Boffenberg K. Substitution von benzol durch elementares Fluor in der Gasphase. –Chem.Ztg.,1972, v.96, № 2, p.84-92.
37. Patent JP 58-41829. Poluchenie oktaftorpropana./ Fukui Siro, Joneda Hadzime. –Publ. 11.03.83, RZh Khimiya, 1984, 5N11.
38. Hayes L.J., Dixon D.D. Direct fluorination of polyester and related compounds. –J.Fluor.Chem., 1977, v.10, № 1, p.1-16.
39. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropolyethers by direct fluorination: a novel preparation for perfluoro (polypropylene oxide) ethers and perfluoro (polymethylene oxide) ethers. –J.Chem.Soc., 1981, part 1, № 5, p.1321-1328.
40. Patent US 4113772. Metod polucheniya oligomerov perftorehfirov s koncevymi karboksil'nymi gruppami. / Lagov R.J. –Publ. 12.09.78, RZh Khimiya, 1979, 15N14.
41. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropoly (ethylene glycol) ethers by direct fluorination. –J.Org.Chem., 1978, v.43, № 23, p.4505-4509.
42. Adcock J.L., Znoue Shoji, Lagow R.J. Simultaneous fluorination and functionalization of hydrocarbon polymers. –J.Amer.Chem., 1978, v.100, № 6, p.1948-1950.
43. Lagow R.J., Margrave J.L. The controlled reaction of hydrocarbopolymers with elemental fluorine. –J.Polym.Sci.: Polym.Lett.Ed., 1974, v.12, № 4, p.177-184.
44. Patent US 3775489. Process for fluorination of aromatic and polynuclear hydrocarbon compounds and fluorocarbons produced thereby. Margrave J.L., Lagow R.J. –Publ. 27.11.73.
45. Lagow R.J., Maraschin N.J. Direct fluorination of cyclic and bicyclic hydrocarbons. –7th Int.Symp.Fluorine Chem., Santa Cruz, Calif., 1973, s.l., s.a., p.1-25.
46. Gerhardt G.E., Dumitru E.T., Lagow R.J. Synthesis of hightbranched perfluoroethers by direct fluorination, promising new materials based on the hexafluoroacetone –ethylene copolymer. –J.Polym.Sci., 1980, v.18, № 1, p.157-169.
47. Robertson G., Liu E.K.S., Lagow R.J. Synthesis of perfluoroadamantane compounds by direct fluorination. –J.Org.Chem., 1978, v.43, № 26, p.4981-4983.
48. Lagow R.J. Large scale synthesis of organofluorine compounds using elemental fluorine; a third Simons cell. –Int.Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.2.
49. Adcock J.L., Lagow R.J. The synthesis of the fluorinated ethers “perfluoroglyme” and “perfluorodiglyme” by direct fluorination. –J.Org.Chem., 1973, v.38, № 20, p.3617-3618.
50. Koshar R.J., Hausted D.R., Meiklejohn R.A. Organic fluoronitrogens. V. Bis(difluoroamino)difluoromethane. –
J.Org.Chem., 1966, v.31, № 12, p.4232-4234.
51. Koshar R.J., Hausted D.R., Wright C.D. Organic fluoronitrogens. VII. Tris(difluoroamino)fluoromethane and related compounds. –J.Org.Chem., 1967, v.32, № 12, p.3859-3864.
52. Patent US 3981783. Process ehlektrohimicheskogo ftorirovaniya s dopolnitel'noj podachej vodoroda i uvelicheniem vyhoda po toku. / Chailds V.V. –Publ.21.09.76, RZh Khimiya, 1977, 14L237.
53. Patent DE 2106870. Sposob ehlektrohimicheskogo ftorirovaniya organicheskih soedinenij./Voss P., Niderprum H., Kaul G., Trepp R., -Publ. 24.02.77, RZh Khimiya, 1977, 24N16.
54. Patent DE 2302132, 1976. Sposob polucheniya razvetvlyonnyh perftoralkanov. /Benninger S. -Publ. 23.12.76, RZh Khimiya, 1977, 24L202.
55. Patent US 4035250. Sposob polucheniya perftorgeptana. /Valters H.S., Chailds V.V. -Opubl.12.07.77, RZh Khimiya, 1978, 6N22.
56. Patent JP 53-18488. Sposob polucheniya perftorcikloalkanov. /Sato Dajsuke, Yamamuti Koiti, Murasima Ryoshiro. -Opubl. 15.06.78, RZh Khimiya, 1979, 9N121.
57. Patent US 3662009. Poluchenie nenasyshchennyh ftorsoderzhashchih soedinenij. /Hadchinson V.M. -Publ. 09.05.72, RZh Khimiya, 1973, 9N12.
58. Goldshtejn B.V., Serushkin I.L., Nikonorova N.I. O roli ftorida nikelya v reakciyah ehlektrohimicheskih soedinenij. –Tr. GIPKh, № 39, p. 1, inv. № T-1863., L., 1975, p.43-47.
59. Fauler R.D., Berford N.B., Gamilton J.M., Sweet R.J., Weber K.E., Kasper J.S. Laitain N. Sintez ftoruglerodov. «Himiya ftora», v. № 1 –M.: IL, 1950, p.91-113.
60. Benner R.J., Benning A.F., Downing F.B., Irvin S.F., Jonson K.S., Linch A.L., Parmeli H.M., Wirt W.W. Ftoruglerody, poluchennye ftorirovaniem uglevodorodov trekhftoristym kobal'tom. «Himiya ftora», v. № 1 –M.: IL, 1950, p.114-128.
61. Fauler R.D., Anderson H.S., Gamilton J.N., Berford N.B., Spagetti A., Biterdih S.B., Laitain N. Ftoridy metallov, primenyaemye v sinteze ftoruglerodov. V kn. «Himiya ftora», sb. № 1 –M.:IL, 1950, s.143-153.
62. Patent JP 60-81134. Poluchenie oktaftorpropana. /Katamura Koiti, Kagehyama Yutaka, Nakayama Hidehtosi. –Publ. 09.05.85, RZh Khimiya, 1986, 12N25.
63. Patent JP 60-10933. Poluchenie geksaftorehtana./ Katamura Koiti, Kagehyama Yutaka, Nakayama Hidehtosi. –Publ. 15.06.85, RZh Khimiya, 1986, 11N21.
64. Pak V., Peka J., Cermak V., Sykora F., Petrzila V. Poloprovozni zarizeni pro vyrobu perfluororganickych latek.
–Chem. Prum., 1978, v.28, № 9, p.467-470.
65. Patent FR 2381732. Usovershenstvovannyj sposob perftorirovaniya ciklicheskih uglevodorodov. / Moore R.E. –
Publ. 27.10.78, Izobreteniya v SSSR i za rubezhom, 1979, v.55, № 5, p.67.
66. Patent US 4143079. Sposob polucheniya perftor-1-metil-4-izopropilciklogeksana iz pinena. / Moore R.E. –Publ. 06.03.79, Izobreteniya v SSSR i za rubezhom, 1979, v.55, № 21, p.20.
67. Patent GB 1597914. Sposob perftorirovaniya ciklicheskih uglevodorodov. / Santehch inkorp. –Publ. 16.09.81, Izobreteniya v SSSR i za rubezhom, 1982, v.57, № 12, p.64.
68. Burdon J. The exhaustive fluorination of aliphatic compounds.
–Int. Symp. “Cent. ...”, Paris, 1936, p.9.
69. Moore R. E., Driscoll G. Perfluorination of bicyclic and tricyclic hydrocarbons. –IV-th Winter Fluorine Conf., Abstr, Daytona Bearen, 1979, p.8.
70. Asovich V.S., Prokudin I.P. Osobennosti ftorirovaniya organicheskih veshchestv chetyrekhftoristym ceriem. –Tr. GIPH, № 40, inv. № T-2117, L., 1976, p.25-31.
71. Isikava N., Kobayasi Yo. Ftor. Himiya i primenenie. –
M.: Mir, 1982, p.91.
72. Keidi J.H., Grosse A.V., Barber E.J., Berger L.L., Sheldon Z.D., Poluchenie ftoruglerodov kataliticheskim ftorirovaniem uglevodorodov. V kn. «Himiya ftora», v. № 1 –M.: IL., 1950, p.129-135.
73. Bigelou L.A. Dejstvie ehlementarnogo ftora na organicheskie soedineniya. «Ftor i ego soedineniya (pod red. J. Saimonsa), t.1 –M.: IL., 1953, p.314-335.
74. Musgrave W. K. R., Smith F. Organic Fluorides. PartІІ. The effect of metals on the fluorunationof hydrocarbons. –J. Chem. Soc., 1949, p.3026-3028.
75. Brik T. J. Ftoruglerody; ih svojstva i proizvodstvo vo vremya vojny. V kn. «Ftor i ego soedineniya» (pod red. Saimonsa J.), t.І –M.: IL, 1953, p.355-388.
76. Maraschin N. J., Catsikis B. D., Davis L. H., Larvinen J., Lagov R. J. Synthesis of structurally unusual fluorocarbons by direct fluorocarbon. –J.Am. Chem. Soc., 1975, v. 97, № 3, p.513-517.
77. Patent US 4113453. Apparat dlya nizkotemperaturnogo pryamogo ftorirovaniya. / Lagov R.J, Adkok I.L., Marashin N.I. –Publ. 12.09.78, RZh Khimiya, 1979, 19I130.
78. Patent US 4281119. Apparat dlya pryamogo ftorirovaniya s kontroliruemym ohlazhdeniem po zonam. / Lagov R.J., Adkok I.L., Marashin N.I. –Publ. 28.07.81, RZh Khimiya, 1982, 9N225.
79. Rahimov A.I., Himiya i tekhnologiya ftororganicheskih soedinenij. –M.: Khimiya, 1986, p.9-10.
80. Schmeisser M., Ehlers K. P., Sartori P. Dichlorhexafluorpropan durch fluorierung von 1,2-dichlorpropan. –Angew. Chem., 1967, v.79, № 13, p.622.
81. Margrave W. K. R., Smith F. Organic Fluorides. PartІ. Fluorination of hydrocarbons. –J. Chem. Sоc., November, 1949, p.3021-3026.
82. Grosse A.V., Keidi J. H. Svojstva ftoruglerodov. V kn. «Himiya ftora»: № 1 –M.: IL, 1950, p.35-64.
83. Patent GB 1281822. Improved Fluorination Process. / Kingdom R. J., Bond G. D. –Publ. 19.07.72.
84. Hill M. Process and market development of fluorocarbon fluids. –Chem.аnd Ind., 1975, № 3, p.118-121.
85. Patent FR 2028457 (V). Sposob ftorirovaniya. / Imperial Smelting Korp. –Publ. 15.01.70, Off. Bull. Francii, Himiya i metallurgiya, 1970, № 45-48, v.І, p.44.
86. Patent US 4220606. Sposob polucheniya perftorproizvodnyh iz ciklichekih uglevodorodov. / Moore R.E. –Publ. 02.09.80, RZh Khimiya, 1981, 9N105.
87. Patent US 3480667. Method of producing fluorinated compounds. / Siegart W. R., Blackley W. D. –Publ. 25.11.69.
88. Patent US 4330475. Aehrozol'nyj sposob pryamogo ftorirovaniya i ustrojstvo dlya ehtoj celi. / Adkok D.L., Renk E.B. –Publ. 18.05.82, RZh Khimiya, 1983,ІІN76.
89. Ruff J. K. The catalytic fluorination of perfluorocarbon nitriles and imines. –J. Org. Chem., 1967, v.32, № 5, p.1675-1677.
90. Lusting M., Ruff J. K. Fluorination of some perfluoro alkyliminosulfurdifluorides. –Inorg. Chem., 1965, v.4, № 10, p.1444-1446.
91. Fokin A.V., Stolyarov V.P., Radchenko V.P. Sintezpoliftoramino-iα-ftornitrosoedinenij na osnove reakcij gazoobraznogo ftora. –Izv. AN SSSR, ser. khim., 1982, № 8, p.1853-1861.
92. Fokin A.V., Galahov V.S., Uzun A.T. i dr. Reakciya ftora s solyami shchelochnyh metallov dinitroacetonitrila v prisutstvii ftoridov kaliya ili kal'ciya. –Izv. AN SSSR, ser. khim., 1974, № 2, p.456-458.
93. Fokin A.V., Uzun A.T., Stolyarov V.P. Bis (diftoramino) ftoracetal'degid –novyj predstavitel'α,α-bis (diftoramino) al'degidov. –Izv. AN SSSR, Ser. khim., 1982, № 6, p.1438.
94. Eremenko L.T., Nacibullin F.YA., Borovinskaya I.P., Karpova N.D. Sintez perftornitroehtanov. –Izv.AN SSSR, Ser.khim., 1968, № 3,p.431-432.
95. Sekiya A., Des Martean D. D. Synthesis of 1,1-bis (fluoroxy) –perhaloalkanes by reaction of fluorinated carboxylic acids with fluorine in the presence of cesium fluoride. –Inorg. Chem., 1980, v. 19, № 5, p.1328-1330.
96. Lu S., Des Martean D. D. Direct synthesis of fluorinated peroxides. 7. Perfluoro-tret-butyl-fluoroformyl peroxide. –Inorg. Chem., 1978, v. 17, № 2, p.304-306.
97. Schack C. J. A new synthesis of difluoraminotrifluoromethane. –J. Fluor. Chem., 1981, v. 18, no 4, p.583-586.
98. Patent DE 2712732. Sposob polucheniya oktaftorpropana. /Halaz S.P. –Publ. 28.09.75, RZh Khimiya, 1979, 14N14.
99. Patent US 4158023. Sposob polucheniya oktaftorpropana. / Halaz S.P. –Opubl. 12.06.79. Izobreteniya v SSSR i za rubezhom, 1979, v. 55, № 24, p.121.
100. Patent GB 1568020.Sposob polucheniya oktaftorpropana. / Halaz S.P. –Publ. 21.05.80. Izobreteniya v SSSR i za rubezhom, 1981, v. 55, № 3, p.54.
101. Patent US 4377715. Poluchenie perftorpropana. /Nuchka H.R., Hino I.B., Abek R.E., Robinson N.A. –Publ. 22.03.83, Izobreteniya v SSSR i za rubezhom, 1983, v.57, № 23, p.88.
102. Patent EU 0031519. Sposob ftorirovaniya organicheskih soedinenij ehlementarnym ftorom. / Ellaid Chemical Co. –Publ. 08.07.81, Izobreteniya v SSSR i za rubezhom, 1983, v.57, № 1, p.20.
103. Patent EU 0032210. Sposob ftorirovaniya organicheskih soedinenij ftorom v trubchatom reaktore iz poristogo metalla v prisutstvii perftorirovannogo razbavitelya. / Ellaid Chemical Co. –Publ. 22.07.81, Izobreteniya v SSSR i za rubezhom, 1983, v.57, № 2, p.27.
104. Patent US 4513154. Sposob provedeniya posledovatel'no-konkuriruyushchih gazofaznyh reakcij. / Kurtz B.E. –Publ. 23.04.85, RZh Khimiya, 1986, 4N25.
105. Patent US 2831035. Proizvodstvo ftorirovannyh uglerodov. / Tuchkovskij E.A., Wolf C. –Publ. 15.04.58, RZh Khimiya, 1960, 85740.
106. Patent US 3709800. Poluchenie perftorirovannyh uglevodorodov./ Fox H.M. –Publ. 09.01.73, RZh Khimiya, 1973, 22N23.
107. Патент Японии 5608. Фторсодержащие галоидуглеводороды. / Осиба Такаси. –Опубл. 22.03.65, РЖ Химия, 1968, 5Н35.
108. Patent US 2681267. Process uluchsheniya kataliticheskih svojstv ftoristogo alyuminiya i produktov iz nego. / Kalfee J. D., Miller Ch.B. –Publ. 15.06.54, RZh Khimiya, 1956, № 14, 45553.
109. Hohorst F. A., Shreeve J. M. Bis-(fluoroxy)-difluoromethane,CF2(OF)2. –J. Am. Chem. Soc., 1967, v.89, № 8, p.1809-1819.
110. Walker N., Des Martean D. D. Direct Synthesis of fluorocarbon peroxides.ІІІ. The addition of chloroperoxytrifluoromethane to olefins. –J. Am. Chem.Soc., 1975, v.97, № 1, p.13-17.
111. Patent US 4499024. Nepreryvnyj sposob polucheniya bis (ftoroksi) diftormetana. / Filolt M.G. –Publ. 12.02.85, RZh Khimiya, 1985, 20N14.
112. Lusting M. A., Pitochelli A. R., Ruff J. K. The catalytic addition of fluorine to a carbonyl group. Preparation of fluoroxy compounds. –J. Am. Chem. Soc., 1967, v. 89, № 12, p.2841-2843.
113. Ruff J. K., Pitochelli A. R., Lusting M. A. A simple synthesis of fluoroxyperfluoroalkyl compounds. -J. Am. Chem. Soc., 1966, v.88,№ 19, p.4531-4532.
114. Kennedy R. C., Cady G. H. Reaction of carbonyl fluoride with fluorine in the presence of various fluorides as catalysts. –J. Fluor. Chem., 1973, v.3, № 1, p.141-154.
115. Muhametshin F. M. Uspekhi himii ftororganicheskih gipogalogenitov i rodstvennyh soedinenij. –Uspekhi khimii, 1980, t. 49, № 7, p.1260-1288.
116. Muhametshin F.M. Gipoftority i ih primenenie v organicheskom sinteze. –V kn.«Novye ftoriruyushchie reagenty v organicheskom sinteze». –Novosibirsk: Nauka, 1987,p.140-196.
117. Cady G. H. Proposed mechanisms of catalysis of fluorination of CF2O. –Anales. Asoc. Quim. Argentina, 1971, v.59, p.3-4, p.125-131.
118. Patent US 3230264. Reaction of carbonyl fluoride with fluorine. –Roger S. P., Cady G.H. –Publ. 18.01.66.
119. Wechsberg M., Cady G.H. Comparative studies of catalytic fluorination of carbon monoxide with elementary fluorine. –J. Am. Chem. Soc., 1969, v.91, p.4432-4435.
120. Kellog K. B., Cady G.H. Trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1948, v.70,№ 12, p.3986-3988.
121. Gervasi J.A., Brown M., Bigelow L. A. The action of elemental fluorine upon organic compounds. XX. The fluorination of mono-, di-and trimethylamine, ethylenediamine and ethyleneimine. –J. Am. Chem. Soc., 1956, v.78, № 8, p.1679-1682.
122. Avonda F. P., Gervasi J. A., Bigelow L.A. The action of elemental fluorine upon organic compounds. XXІ. The fluorination of malononitrile and dimethylformamide. –J. Am. Chem. Soc., 1956, v.78, № 12, p.2798-2800.
123. Toitelboim M.A., Shoihet A.A., Kaplunov M.G., Vedeneev V.I. Energeticheskie razvetvleniya cepej v reakciyah triftormetilgipoftorita s galoidmetanami. –Kinetika i kataliz, 1981, t. 22, v. 2, p. 298.
124. Toitelboim M.A., Shoihet A.A., Vedeneev V.I. Energeticheskie razvetvleniya cepej v reakciyah triftormetilgipoftorita s galoidmetanami. 2. Mekhanizm otricatel'nogo vzaimodejstviya cepej. –Kinetika i kataliz, 1981, t. 22, v. 3, p. 564-568.
125. Vedeneev V.I., Parijskaya A.V. Mekhanizm ftorirovaniya metana i ego ftorproizvodnyh. І. Sravnenie skorostej ftorirovaniya metana, ftormetana, diftormetana i triftormetana. –Kinetika i kataliz, 1971, t. 12, v. 1, p. 21-26.
126. Parijskaya A.V., Vedeneev V.I. Mekhanizm ftorirovaniya metana i ego proizvodnyh. 2. Diftormetan –Kinetika i kataliz, 1971, t.12, v.2, p.293-298.
127. Parijskaya A.V., Vedeneev V.I. Mekhanizm ftorirovaniya metana i ego ftorproizvodnyh. 3. Ftoristyj metil. –Kinetika i kataliz, 1971, t.12, v.3, p.543-548.
128. Parijskaya A.V., Vedeneev V.I. Mekhanizm ftorirovaniya metana i ego ftorproizvodnyh.4. Metan. –Kinetika i ataliz, 1971, t. 12, v.4,p. 839-842.
129. Nadtochenko V.A., Fedotov N.B., Vedeneev V.I., Sarkisov O.M. O reakcii kolebatel'no vozbuzhdennoj molekuly CH3F s ftorom. –Dokl. AN SSSR, 1978, t.238, № 6, p.1391-1394.
130. Parijskaya A.V., Vedeneev V.I. O prirode zaderzhek samovosplameneniya v sisteme CH3F + F2 + O2 + He. –Kinetika i kataliz, t.14, v.6, p.1365-1369.
131. Vedeneev V.I., Teitelboim M.A., Shoihet A.A. Fotohimicheskoe ftorirovanie ftoroforma v prisutstvii kisloroda. –Izv. AN SSSR, 1976, № 9, p.1968-1970.
132. Medvedev B.A., Teitelboim M.A., Shilov A.E. Himicheskaya aktivaciya molekuly CHFCl2 v reakcii CHCl2+ F2→ CHCl2F + F . –Kinetika i kataliz, t.12, v.2, p. 269-275.
133. Medvedev B.A., Teitelboim M.A., Shilov A.E. Mekhanizm gazofaznoj reakcii hlorftormetana s molekulyarnym ftorom. –Kinetika i kataliz, 1971, t.12, v.3, p. 49-751.
134. Vedeneev V.I., Medvedev B.A., Teitelboim M.A. Gazofaznoe ftorirovanie diftormetana pri povyshennyh davleniyah inertnogo gaza. –Kinetika i kataliz, t.13, v.1, p.50-53.
135. Obvivalneva A.A., Fedotov V.G. Vozbuzhdennye molekuly v reakcii ftora s acetonom (ftoracetony; sintez; mekhanizm). –Kinetika i kataliz, 1981, t.22, v.5, v.1095-1099.
136. Kapralova G.A., Margolina E.M., Rusin L.Y., Chajkin A.M., Shilov A.E. Rol' kolebatel'no-vozbuzhdennyh molekul v reakciyah ftorirovaniya molekulyarnym ftorom. –V sb. «5 Mezhdunar. simpozium po himii ftora», Tezisy dokl., M., : Nauka, 1969, p.102-104.
137. Knunyats I.L, Gambaryan N.P., Rohlin E.M. Karbeny. (Soedineniya dvuhvalentnogo ugleroda, promezhutochno obrazuyushchiesya v organicheskih reakciyah). –Uspekhi khimii, 1958, t.27, v.12, s.1361-1436.
138. Muhametshin F.M., Zhirnov O.M., Ainagos N.A., Petrov Y.I., Lyapunov M.I. O nekotoryh osobennostyah gipoftoritnogo metoda polucheniya perftormetilvinilovogo ehfira. Soobshch. 2. Sintez triftormetilgipoftorita. –Tr. GIPH, № 69, inv. № T-2769, L., 1980, p.73-78.
139. Muhametshin F.M., Povroznik S.V., Zhirnov O.M. Issledovanie kinetiki i mekhanizma kataliticheskoj reakcii obrazovaniya triftormetilgipoftorita. –Tr. GIPH, № 103, inv. № T-2981, L., 1983, p.75-80.
140. Patent SU 1141707. Sposob polucheniya 1,1-diftor-1-hlorehtana ili 1,1,2,2-tetraftordihlorehtana. /Zaharov V.Y., Goldinov A.L., Borovnev L. M., Golubev A.N., Kalashnikova N.A., Zaharova O.M., Lyubimova L.A.
141. Kapralova G.A., Buben S.N., Chaikin A.M. Kinetika i mekhanizm reakcii ftorirovaniya okisi ugleroda. –Kinetika i kataliz, 1975, t.16, v.3, p.591-595.
142. Povroznik S.V. Razrabotka sposoba i tekhnologii polucheniya ftoroksitriftormetana reakciyami ehlementarnogo ftora s karbonilsoderzhashchimi soedineniyami. Diss. kand. tekhn. nauk, 1987, PF NPO GIPH, 161p.
143. Krasnov K.S. Molekuly i himicheskaya svyaz'. –M.: Vyssh. shkola, 1977, p.117.
144. Bezmelicyn V.N., Legasov V.A., Chaivanov B.B. Sintez diftorida kriptona s primeneniem termicheskoi generacii potokov atomarnogo ftora. –Dokl. AN SSSR. Ser. khim., 1977, t.235, № 1, p.96-98.
145. Bezmelicyn V.N., Legasov V.A., Spirin S.N., Chajvanov B.B. Kinetika kataliticheskoi atomizacii ftora. –Dokl. AN SSSR. Ser.fiz.khimiya, 1982, t.262, № 5, p.1153-1157.
146. Palkina L.A., Spirin S.N., Tishchenko P.P. processy s uchastiem atomarnogo ftora. –Dokl. AN SSSR. Ser.fiz.khimiya, 1982, t.262, p.1428-1433.
147. Bezmelicyn V.N., Vasilev A.A., Sinyanskij V.F., Chajvanov B.B. Kinetika termokataliticheskoj dissociacii ftora na poverhnosti nikelya. –Tezisy dokladov na 8 Vsesoyuzn. simp. po himii neorg. ftoridov, 1987, M.: Nauka, p.59.
148. Nikonorov YU.I. Ftoridy nikelya. Ih kataliticheskaya i okislitel'naya aktivnost'. –Tezisy dokladov na 5 Vsesoyuzn. simp. po himii neorg. ftoridov, 1978, M.: Nauka, p.205.
149. Ryss I.G. Khimiya ftora i ego neorganicheskih soedinenij. –M.: Goskhimizdat, 1956, p.576-579.
150. Redwood M.E., Willis C. J. Fully fluorinated alkoxides. PartІ. Trifluoromethoxides of alkali metals. –Canad. J. Chem., 1965, v.43, p.1893-1898.
151. Gubanov V.A., Veretennikov N.V., Troichanskaya P.E., Dolgopol'skij I.M. Sintez perftoralkoksidov metallovІ gruppy i termograficheskoe izuchenie ih svojstv. –Zh. Org. khim., 1975, t.ІІ, № 2, p.322-325.
152. Levy J. B., Kennedy R. C. Bis (trifluoromethyl) peroxide.І. Thermodynamics of the equilibrium with carbonyl fluoride and trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1972, v.94, no 10, p.3302-3305.
153. Patent JP 35888. Sposob polucheniya triftormetilgipoftorita. / Nagase Syundzi, Abe Takasi, Baba Hadzime, Ohira Kadzuo. –Publ. 09.09.72, RZh Khimiya, 1973, 14N120.
154. Patent US 3687825. Sposob polucheniya triftormetilgipoftorita. Nagase Syundzi, Abe Takasi, Baba Nadzime. –Publ. 29.08.72, RZh Khimiya, 1973, 21N26.
155. Patent US 2983764. Sposob polucheniya ftoruglerodnyh soedinenij./ Knak D.F. –Publ. 9.05.61, RZh Khimiya, 1962, 14L53.
156. Patent JP 56-16429. Oligomerizaciya geksaftorpropilena v gazovoj faze. / Osaka Yonosuke, Higasidzuka Takehsi. –Publ. 17.02.81, RZh Khimiya, 1982, 3N10.
157. Patent JP 50-45818. Poluchenie oligomerov geksaftorpropilena. Kadzava Masahiro, Komatsu Tadaaki, Macuoka Kimiaki. –Publ. 08.06.81, RZh Khimiya, 1982, 9NІІ.
158. Patent US 4296265. Sposob polucheniya oligomerov geksaftorpropilena. / Osaka Yonosuke, Tozuka Takasi. –Publ. 20.10.81, RZh Khimiya, 1982, І3NІІ.
159. Patent DE 3027229. Sposob polucheniya oligomerov geksaftorpropilena. / Osaka Y., Tozuka T. –Publ. 07.02.81, Izobreteniya v SSSR i za rubezhom, 1981, V.55, № 12, p.30.
160. Patent BE 2055824A. Sposob polucheniya oligomerov geksaftorpropilena. /Osaka Y., Tozuka T. –Publ. 18.07.80, Sbornik referatov NIOKR, obzorov, perevodov i dep. rukopisej. DSP. Khimiya i khimicheskaya tekhnologiya, M., 1986, № 5, p.14.
161. Patent DE 2616733. Sposob polucheniya di- i trimerov geksaftorpropilena. / Okava M., Komatsu T., Matsuoka K. –Publ. 20.03.80, Izobreteniya v SSSR i za rubezhom, 1980, № 15, p.13.
162. Patent JP 57-2697. Sposob polucheniya oligomerov geksaftorpropena. / Neosu K.K. –Publ. 18.01.82, Izobreteniya v SSSR i za rubezhom, 1982, v.57, № 14, p.113.
163. Scherer K. V., Ono T., Jamanouchi K., Fernandez R., Henderson P.B. F-2,4-Dimethyl-3-ethyl-3-pentyl and F-2,4-dimethyl-3-isopropyl-3pentyl: stable tert-perfluoroalkyl radicals prepared by addition of fluorine or trifluoromethyl to a perfluoroalkene. –J. Am. Chem. Soc., 1985, v.107, № 3, p.718-719.
164. Scherer K. V., Fernandez R., Henderson P.B. Long lived perfluoroalkyl radicals: preparation by direct fluorination and alternate routes, properties and rates andmechanisms of disappearance. –Int. Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.1.
165. Patent DE 2332097. Sposob polucheniya perftornonanov. /Halaz S.P. –Publ. 16.01. 75, RZh Khimiya, 1975, 18N25.
166. Patent DE 2332088. Sposob polucheniya perftor-2-metilpentana./ Halaz S.P. –Publ. 16.07.81, Izobreteniya v SSSR i zarubezhom, 1981, v.55, № 23, p.21.
167. Allayarov S.R., Barkalov I.M., Gol'danskij V.I., Kiryuhin D.P. Obrazovanie stabil'nyh radikalov iz ftororganicheskih soedinenij. –Izv. AN SSSR. Ser. him., 1983, № 6, p.1225-1228.
168. Allayarov S.R., Barkalov I.M., Gol'danskij V.I., Demidov S.V., Kiryuhin D.P., Mihajlov A.I. Stabil'nye radikaly –rastvorennye perftoralkily. –Izv. AN SSSR. Ser. him., 1983, № 6, p.1448-1449.
169. Allayarov S.R., Mihailov A.I., Barkalov I.M. Novyj stabil'nyj perftoralkil'nyj radikal. –Izv. AN SSSR. Ser. him., 1985, № 7, p.1667-1669.
170. Allayarov S.R., Barkalov I.M., Lebedov M.YU., Loginova N.N., Mihailov A.I. Obrazovanie stabil'nyh radikalov pri radiolize geksaftorpropilena. –Izv.AN SSSR. Ser. him., 1986, № 2,p.462-463.
171. Allayarov S.R., Barkalov I.M., Gol'danskij V.I. Obrazovanie dolgozhivushchih radikalov pri ftorirovanii zhidkih uglevodorodov. – Izv.AN SSSR. Ser. him., 1986, № 10, p.394.
172. Allayarov S.R., Sumina I.V., Barkalov I.M., Mihailov A.I. Obrazovanie stabil'nyh radikalov pri fotolize perftor-2,4-dimetil-3-ehtilpentena-2. –Izv. AN SSSR, Ser. him., 1986, № 10, p.2359-2361.
173. Krusic P. J., Scherer K. V. An electron spin resonance study of long lived fluoroalkyl radicals generated by photolysis of hindered perfluoroalkenes. Int. Symp. “Centenary of the discovery of fluorine”. Abstr., Paris, 1986, p.37.
174. Levy J.B., Kennedy R. C. Homolytic displacement reactions of carbon.І. The fluorine-perfluorocyclobutane reactions. –J.Am.Chem.Soc., 1974, v.96, № 15, p.4791-9795.
175. Gol'dinov A.L., Zaharov V.Y., Baibakov P.Y., ZHukova V.A., Novikova M.D. Novye aspekty pryamogo ftorirovaniya. Kataliticheskoe gazofaznoe ftorirovanie ftorolefinov; poluchenie perftoruglerodov. –Otchet predpriyatiya p/ya A-1619, 1987, inv. № 1288, 87p.
176. Matveev S.A., Prokudin L.P., Timofeev N.V. Ispol'zovanie triftorida kobal'ta dlya ftorirovaniya organicheskih soedinenij po dvojnym svyazyam. –Tr. GIPH, 1975, № 31, inv. № T-1728, p.119-127.
177. Asovich V.S., Prokudin I.P. Ftorirovanie acetilena ftoridami kobal'ta i ceriya. Tr. GIPH, № 55, inv. № T-2552, L., 1978, p.21-24.
178. Asovich V.S., Prokudin I.P., Maksimov B.N., Stepanov V.P. Izuchenie kineticheskih zakonomernostej ftorirovaniya acetilena trekhftoristym kobal'tom. –Tr. GIPH, № 65, inv. № T-2723, L., 1979, p.42-47.
179. Kotikov S.V., Prokudin I.P., Asovich V.S. Izuchenie kineticheskih zakonomernostej ftorirovaniya geksaftorpropilena trekhftoristym kobal'tom. –Tr. GIPH, № 101, inv. № T-2986, L., GIPH, 1985, s.24-27.
180. Burdon J., Knights J.R., Parsons J. W., Tatlow J. C. The fluorination of ethane and ethene over potassium tetrafluorocobaltate (ІІІ) and cobalt trifluoride. –Tetrahedron, 1976, v.32, p.1041-1043.
181. Gol'dinov A.L., Borovnev L.M., SHirokova N.S., Kovbasyuk O.G., Tishina V.V. Vliyanie tekhnologicheskih parametrov sinteza i chistoty rastvoritelya na svojstva ftoroplasta-50. –Otchet predpriyatiya p/ya A-1619, 1987, inv. № 1231.
182. Patent DE 2451493. Sposob polucheniya perftorirovannyh ehfirov./ Halaz S.P., Klag F. –Publ. 06.05.76, Izobreteniya v SSSR i za rubezhom,1982, v.57, № 22, p.5.
183. Patent US 3665041. Perfluorinated polyethers and process for their preparation. / Sianesi D., Fontanelli R., -Publ. 23.05.72.
184. Ryabinin N.A., Dolubenov A.E., Shmatov L.E. Ftorpoliehfiry kak antiadgezivy pri pressovanii perhlorata nitroniya. –Tr. GIPH, № 101, inv. № T-2986, L., 1985, p.83-87.
185. Patent SU 1332758. Sposob ochistki geksaftorpropilena, tetraftorehtilena i ih semej ot oktaftorizobutilena. /Fokin A.V., Studnev Y.N., Rapkin A.I. i dr.
186. Gol'dinov A.L., Borovnev L.M., Golubev A.N., Zaharov V.Yu. i dr. Razrabotka tekhnologii polucheniya 2-perftormetil-4-oksaperftornonana na osnove perftorizobutilena i 1,1,5-trigidroperftor pentanola. –Otchet predpriyatiya p/ya A-1619, 1987, inv. № 1229, 39p.
187. Patent SU 1148285. 3-Gidropropilftorsul'fat v kachestve promezhutochnogo produkta dlya sinteza 2,2,3,3-tetraftorpropionovoj kisloty i sposob ee polucheniya. /Fokin A.V., Studnev Y.N., Rapkin A.I. i dr.
188. Patent SU 3966753. Sposob polucheniya 2-gidrotetraftorehtilftorsul'fata. /Fokin A.V., Rapkin A.I., Tatarinov A.S. i dr.
189. Patent SU 1233595. Sposob polucheniya ω-gidroperftorkarbonovyh kislot (ego varianty). / Fokin A.V., Studnev Y.N., Rapkin A.I. i dr.
190. Tekhnicheskie trebovaniya k PFDMCG dlya ispol'zovaniya v radioehlektronike. M.: INEOS AN SSSR. Reg. № 12111-10 DSP ot 18.01.85.
191. Iskhodnye dannye dlya razrabotki zhidkosti-diehlektrika. M.: INEOS AN SSSR. Pril. k vh.№ 155 ot 18.02.85.
192. Gol'dinov A.L., Zaharov V.YU., Bajbakov P.YA. i dr. Razrabotka tekhnologii polucheniya perftordimetilciklogeksana povyshennoj chistoty. –Otchet predpriyatiya p/ya A-1619, 1986, inv. № 1218, 24p.
193. Gol'dinov A.L., Maslyakov A.I., Zaharov V.Y. i dr. O vozmozhnosti rezkogo uvelicheniya vypuska deficitnyh ftoruglerodnyh zhidkostej i smazok. –Otchet predpriyatiya p/ya A-1619, 1986, № 1219, 13p.
194. Vremennyj tekhnologicheskij reglament proizvodstva perftordekalina.1981, inv. № 1115.
195. Furin G.G. Synthetic aspects of the fluorination of organic compounds. (Ed.) M.E. Vol’pin./Harwood Academic
Publishers GmbH: United Kingdom, Sov. Sci. Rev., B. Chem., 1991, V.16, Part 1,р.140.
196. Rozen S., Kol M. Olefin epoxidation using elemental fluorine. / J.Org.Chem., 1990. V.55,р.5155-5159.
197. Patent JP 02-131438, (1990). CO719/08. Preparation of hexafluoroethane. /Shimuzu M.C.A. 1990, V.I 13,97031.
198. Patent JP 62-193946, (1989). CO719/08, CO17/04. Sposob polucheniya ftorsoderzhashchih ehtanov. /Faruka Y., Kono H. RZh Kh, 1990, 2N 131.
199. Lagow R.J. Important new developments in direct fluorination technology. /Memorial symposium for Prof. Nobuo Ishikawa, Dec. 9-10, 1991, Nippon Kaiun Club, Japan.
200. Lin W.H., Lagow R.J. Synthesis of perfluorodicyclohexano-crown-6 ether. /J.Chem.Soc., Chem.Commun., 1991,р.13-14.
201. Clark W.D., Lin T.Y., Maleknia S.D., Lagow R.J. Synthesis of perfluorotetralkyl ortocarbonates using elemental fluorine. / J.Org.Chem., 1989. V.54.р.1990-1992.
202. Sievert A.C., Tong W.R., Nappa M.J. Preparation D’analogues fluores de la pheromone d’anthanomus grandis. / J.Fluorine Chem., 1991. V.51.р.397-405.
203. Tonelli C., Tortelli V. Photoinduced fluorination of hexafluoropropene trimers: synthesis of branched perfluoroalkanes. /J.Chem.Soc., Perkin Trans I, 1990, № 1,р.23-26.
204. Patent Application EP 344935, (1989). CO8G65/00. Preparation of prefluoroethers by photo-assisted fluorination. / Nappa M.J., Sievert A. C.A. 1990, V. 112, 216229.
205. Patent US 4960951, (1990). CO743/12. Novel perfluoroalkyl ethers by two-step fluorination of polyols./ Nappa M.J., Sievert A.C. Tong W.R. C.A. 1991, V. 111, 216229.
206. Sievert A.C., Tong W.R., Nappa M.J. Synthesis of perfluorinated ethers by an improved solution phase direct fluorination process. / J.Fluorine Chem. 1991. V. 53,р. 397-417.
207. Mizin G.G., Simakov B.V., Shultezkaya L.V., Moldavsky D.D. Issledovanie sposoba polucheniya perftorehtilciklogeksena. / ZHPH. 1991. t.64 ,v.6. p.293-1296.
208. Moldavsky D.D., Mizin G.G., Shultezkaya L.V., Simakov B.V., Furin G.G. Synthesis of perfluoroethylcyclohexene, its alkoxy and diakylamino derivates and their electrochemical fluorination. / Abstracts of 10-European Symposium on Fluorine Chemistry, Sept. 20-25, 1992, Padua, Italy. Abstracts. B7; J. Fluorine Chem., 1992. v.58.р.185.
209. Moldavsky D. D. Perftororganicheskie soedineniya: Poluchenie pryamym ftorirovaniemuglevodorodov. Diss. dokt. him. nauk, 2002, RNC Prikladnaya himiya, 268p.
210. Suvorov B.V., Bukeihanov N.R. Okislitel'nye reakcii v organicheskom sinteze. 1978g. M., Khimiya, 197p.
211. Semenova G.A., Leites I.L., Aksel'rod Y.V., Markina M.I., Sergeev S.P.,Har'kovskaya E.N. Kataliticheskie i adsorbcionnye metody ochistki gazov ot sernistyh soedinenij. –Ochistka tekhnologicheskih gazov.1977g, M.,Khimiya, 1977,p. 35-37.
212. Panov G.I. Zakonomernosti geterogenno-kataliticheskoj aktivacii dvuhatomnyh molekul (H2, O2, N2). Novye kataliticheskie sistemy aktivacii molekulyarnogo azota. Diss. dokt. him. nauk, 1986, Novosibirsk, 374p.
213. Patent SU 220740. Sposob polucheniya tetrahlortetraftorpropana. –Palleta O., P. Svoboda, L. Stefan, Y. Kvisala. –Publ. 15.12.85. RZhKhimiya, 1986, ІІN24.
214. Patent JP 60-116637. Poluchenie ftormetana. –Takayama S., Meiraku F., Takoiti A., Kovasaki H. –Publ. 24.06.85. RZh Khimiya, 1986, 9N16.
215. Margolis L.Y. Okislenie uglevodorodov na geterogennyh katalizatorah. 1977g. M., Khimiya, p. 296.
216. Kavaev M.M., Zaporin A.P., Kleshchev N.F. Kataliticheskoe okislenie ammiaka. 1983g. M., Khimiya, p. 31.
217. Tekhnologiya katalizatorov. 1974g. L., Himiya, p. 138-140.
218. Patent US 2760997. Process for the production of chlorotrifluoroethylene by passing a mixture of trichlorotrifluoroethane and hydrogen through an unobstructed iron tube. / Rucker J. T., Stormon D. B. –Publ. 28.08.56.
219. Patent US 2615925. Preparation of olefinic compounds. / Bordner C.A. –Publ. 28.10.52.
220. Patent US 2704777. Preparation of halogenated olefines / Clark J.W. –Publ. 22.03.55.
221. Patent GB 698386. Improvement in preparation of halogenated olefines / Clark J. W. –Publ. 14.10.53.
222. Patent DE 1044800. Verfahren zur Herstellung von Chlortrifluorathylen / Clark J. W. –Publ. 27.11.58.
223. Patent US 2685606. Preparation of halogenated olefines / Clark J. W. –Publ. 03.08.54 C.A. 48, 127881 (1954).
224. Patent US 2697124. Dehalogenation of fluorohalocarbons / Russel M.M. –Publ. 14.12.54 C. A. 50, 2650 (1956).
225. Patent US 2864873. Production of 1,1,2-trifluoro-2-chloroethylene / Miller C. B., Smith L. B. –Publ. 16.12.58.
226. Patent US 3043889. Poluchenie ftoristogo ehtilena. / Smith L.B., Wolf C. –Publ. 10.07.62. RZh Khimiya, 1964, 3N24.
227. Patent DE 1186847. Poluchenie 1-chlor-1,2,2-triftorehtilena. / Smit L.B., Wolf C. –Publ. 4.10.65, RZh Khimiya, 1966, 13N26.
228. Patent US 3333011. Poluchenie hlortriftorehtilena./Anello L.G., Wolf C. –Publ. 25.07.67 RZh Khimiya, 1969, 14N30.
229. Patent JP 60-185734. Poluchenie triftorhlorehtilena. / Morimoto Takesi, Morikava Sanesuke, Funayama Keidi. –Publ. 21.09.85, 19N37.
230. Patent EU 0053657. Sposob polucheniya hlortriftorehtilena i triftorehtilena. / Kaningham U.D., Plekors R.E., Smith E.M. –Publ. 16.06.82. Izobr. v SSSR i z/r., vyp. 57, № 15, p. 83.
231. Patent JP 43-8454. Sposob polucheniya triftorehtilena. / Sakata Hirosukeh. –Publ. 02.04.68. RZh Khimiya, 1969, 3N36.
232. Patent US 2913400. Katalizator iz okisi molibdena dlya konversii uglevodorodov. / Burton U.P., Lefrenkois F.A., Riblet E.U. –Publ. 17.11.59, RZh Khimiya, 1961, 9K156.
233. Patent US 3354233. Proizvodstvo tetraftorehtilena. / Anello L.G., Wolf C. –Publ. 21.11.67, RZh Khimiya, 1969, 9N32.
234. Patent DE 1270547. Verfahren zur Herstellung von Tetrafluorathylen/ Anello Z. G., Wolf C. –Publ. 30.01.69.
235. Patent US 3505417. Degaloidirovanie ftorgalogenuglerodov. / Gardner L.E. –Publ. 07.04.70, RZh Khimiya, 1971, 11133.
236. Patent US 3636173. Sposob kataliticheskogo degaloidirovaniya./ Gardner L.E. –Publ. 18.01.72, RZh Khimiya, 1972, 21N21.
237. Patent US 3789016. Katalizator gidrodegaloidirovaniya. / Gardner L.E. –Publ. 29.01.74. RZh Khimiya, 1975, 2N217.
238. Patent US 3636172. Degaloidirovanie ftorgaloiduglevodorodov. / Gardner L.E. –Publ. 18.01.72, RZh Khimiya, 1972, 21N20.
239. Patent US 2900423. Proizvodstvo perftorpropilena. / Smith L.B. –Publ. 18.08.59. RZh Khimiya, 1961, 7L44.
240. Patent US 2734090. Poluchenie ftoristogo vinilidena. / Kafi D.D., Miller CH.B. –Publ. 07.02.56, RZh Khimiya, 1957, 12, 42313.
241. Patent US 3404180. Poluchenie carbonilftorida. / K.K. Lester. –Publ. 1.10.68. –RZh Khimiya, 1969, 23N92.
242. Patent FR 1460246. Sposob polucheniya okisi tetraftorehtilena. / Edison. –Publ. 17.10.66. –RZh Khimiya, 1968ІІNІ24.
243. Sokolov L.F. Issledovanie i razrabotka tekhnologii processov okisleniya ftorolefinov. –Diss. dokt. khim. nauk. –Leningrad, VNIISK, 1979.
244. Patent SU 190555. Sposob polucheniya ftoristogo vodoroda. / Gol'dinov A.L., Romanov E.I., Zaharov V.Y. i dr.
245. Gol'dinov A.L., Borovnev L.M., Zaharov V.Y. i dr. Termicheskoe obezvrezhivanie ftororganicheskih othodov v vodorodo-vozdushnom plameni na opytno-promyshlennoj ustanovke. –Otchet predpriyatiya p/ya A-1619, 1982, inv. № 887, 18p.
246. Borovnev L.M., Golubev A.N., Zaharov V.Y. i dr. Kataliz i iniciirovanie destruktivnogo okisleniya, tetraftorehtilena molekulyarnym kislorodom. Poluchenie karbonilftorida. –Otchet predpriyatiya p/ya A-1619, 1983, inv. № 1110, 21p.
247. Patent DE 213342. Sposob polucheniya ftorsoderzhashchih galoiduglevodorodov so srednej stepen'yu ftorirovaniya. / L. Ingehburg, K. Rehjnfred, M. Diter. –Publ. 24.12.85. –RZh Khimiya, 1986, 21N17.
248. Patent DE 203314. Katalizator polucheniya hlorftormetana v gazovoj faze. / I. Lelid, R. Kaden, D. Mross. –Publ. 19.10.83. –RZh Khimiya, 1984, 17N181.
249. Patent US 4504686. Sposob polucheniya 1,1,2-triftor-2-hlorehtildiftormetilovogo ehfira. / S. Takami. –Opubl. 12.03.85. –RZH Himiya, 1986, 8N9.
250. Patent JP 55-85564. Poluchenie β-triftormetilhlorpiridinov./ R.Nishima, K. Rudzikama, I.Jokomiti, Y. Cudzii, S. Nisimura. –Publ. 27.06.80. –RZh Khimiya, 1981, 19N177.
251. Patent US 4139568. Sposob polucheniya metilftorida. / V. Daniehl, M. Libbs. –Publ. 13.02.79. –RZh Khimiya, 1979, 18N18.
252. Patent US 4145368. Poluchenie 1,1,1-triftor-2,2-dihlorehtana. R. Sweney, S. Bernard. –Publ. 20.03.79. –RZh Khimiya, 1979, 20N20.
253. Patent FR 1453510. Sposob ochistki dihlortetraftorehtana. / Corp. Elektrohimiya. –Publ. 16.08.66. –RZh Khimiya, 1968, 11N39.
254. Vilenchik Y.M., Yakurnova G.I., Ostapenko E.A. Sintez triftoracetilftorida na osnove okisi tetraftorehtilena. –Tr. GIPH, 1985, № 101, p. 147-149.
255. Patent JP 58-35978. Sposob polucheniya pentaftorpropionilftorida. / A. Jonotasi, A. Josio. –Publ. 08.05.83.-Izobreteniya v SSSR i za rubezhom, 1984, v. 57, № 8, part 2, p. 130.
256. Patent JP 58-38231. Poluchenie pentaftorpropionilftorida./ I. Jonosuke, A. Josio, S. Hiroyuki. –Publ. 05.03.83.-RZh Khimiya, 1984, 5N46.
257. Patent US 3250808. Ftorsoderzhashchie prostye ehfiry. / E.F. Mur, A. Mil'yan, G. Eleuterio. –Publ. 10.05.66. –RZh Himiya, 1967, 17N82.
258. Patent US 3250807. Ftorsoderzhashchie karbonovye kisloty s ehfirnymi gruppirovkami. / CH.G. Fritz, E.F. Moore. –Publ. 10.05.66. –RZh Khimiya, 1967, 17N84.
259. Patent US 3242218. Sposob polucheniya ftoruglerodnyh poliehfirov. / V.Miller. –Publ. 22.03.66. –RZh Khimiya, 1967, 12S232.
260. Patent DE 78376. Sposob polimerizacii i sopolimerizacii okisi geksaftorpropilena v prisutstvii geksaftorpropilena. / G. Lothar. –Publ. 12.12.70. –RZh Khimiya, 1971, 21S361.
261. Patent US 3125599. Polimery okisej ftoruglerodnyh soedinenij. / D. Vornell. –Publ. 17.03.64. –RZh Khimiya, 1965, 15S277.
262. Patent US 3291843. Fluorinated vinilethers. / Fritz C.G., Selman S., -Publ. 13.12.66.C.A. 1967, 66, 37427 h.
263. Patent US 4377717. Sposob polucheniya perftor-2-metilpentena-2. / L.Anello, R. Sweeny. –Publ. 22.03.83. –RZh Khimiya, 1983, 24N1311.
264. Knunyats I.L., Fokin A.V., Cheburkov Y.A. Chetvertyi mezhdunarodnyj simpozium po himii ftora v Istes-Park (Kolorado, USA) –ZHVHO im. D.I.Mendeleeva, t. 13, № 3, p. 311-321.
265. Patent US 3449389. Pervichnye ftorzameshchennye alkogolyaty. / D. Vornell. –Publ. 10.06.69. –RZh Khimiya, 1970, 18N39.
266. Patent US 3322826. Polimerizaciya geksaftorpropilenehpoksida./ E.F.Moore –Publ. 0.06.67. –RZh Khimiya, 1968, 18S 296.
267. Patent JP 57-45132. Poluchenie perftor-2-metil-3-oksageksanoilftorida. / Y. Masaki, M. Sehjdzi. –Publ. 13.03.82. –RZh Khimiya, 1983, 5N70.
268. Patent US 3274293. Ftorsoderzhashchie prostye ehfiry. / S.Stanley. –Publ. 20.09.66. –RZh Khimiya, 1968, 5N161.
269. Patent DE 2756919. Sposob demerizacii okisi geksaftorpropilena. / G. Kuhne, F. Geller. –Publ. 05.07.79. –RZh Khimiya, 1980, 14N22.
270. Patent DE 2924385. Sposob dimerizacii okisi geksaftorpropilena. / G. Kuhne. –Publ. 07.08.80. –RZh Khimiya, 1981, 23N36.
271. Kinle H., Bader E. Aktivnye ugli i ih promyshlennoe primenenie. –L., Khimiya, 1984, p. 185-192.
272. Patent US 3321515. Sposob polucheniya ftorirovannyh karbonil'nyh soedinenij. / F.Moore, S.Milyan. –Publ. 23.05.67.-RZh Khimiya, 1968, 15N48.
273. Bekker R.A., Asratyan G.V., Dyatkin B.L. O roli prostranstvennyh faktorov v reakciyah poliftorirovannyhα-okisej s aminami. –ZhOrKh, 1973, t. 9, v. 8, p. 1644-1648.
274. Knunyants I.L., Shokina V.V., Tuleneva, V.V., Ceburkov Y.A., Aronov Y.E. Proizvodnye perftorizomaslyanoj kisloty. –Izv. AN SSSR, ser. khim., 1966, № 10, p. 1831-1833.
275. Sianesi D., Passety A., Tarli F., The Chemistry of hexafluoropropene epoxide. –J. Org. Chem., 1966,v. 31, № 7,p. 2312-2316.
276. Borovnev L.M., Golubev A.N., Zaharov V.Y. i dr. Iniciirovanie processa sopolimerizacii tetraftorehtilena i geksaftorpropilena perftorirovannymi diacil'nymi perekisyami, sintezirovannymi na osnove okisi geksaftorpropilena. –Otchet predpriyatiya p/ya A-1619, 1985, inv. № 1154, 28p.
277. Ashmor. Kataliz i iniciirovanie himicheskih reakcij. M.: Mir, 1966, p. 62.
278. Patent US 3213134. Deciklizaciya ftorirovannyh ciklicheskih efirov. / D. Morin. –Publ. 19.10.65. –RZh Khimiya, 1967, 9N60.
279. Patent US 4302608. Sposob izomerizacii okisi geksaftorpropilena do geksaftoracetona. / E.Skuri, D. Milz. –Publ. 24.11.81. –RZh Khimiya, 1982, 20N56.
280. Patent JP 58-62131. Poluchenie geksaftoracetona. / O. Josio, A. Josimasa, M. Makisuke. –Publ. 13.04.83. –RZh Khimiya, 1984, 12N30.
281. Patent JP 58-62130. Poluchenie geksaftoracetona. / A. Josimasa, M. Makisuke, S. Yukio. –Publ. 13.04.83. –RZh Khimiya, 1984, 12N29.
282. Patent SU 740741. Sposob polucheniya geksaftoracetona. / Y.M. Vilenchik, G.I. Yakurnova, G.I. Lekomceva, L.P. Zayakina, A.P. Harchenko. –Publ. 15.06.80. –RZh Khimiya, 1980, 24N67.
283. Patent US 859348. Sposob polucheniya geksaftoracetona. / Y.M. Vilenchik, G.I. Yakurnova. –Publ. v B.I., 1981, № 32, RZh Khimiya, 1982, 21N12.
284. Patent US 4238416. Sposob izomerizacii ftorirovannyh soedinenij. / D. Koduo. –Publ. 09.12.80. –RZh Khimiya, 1981, 17N65.
285. Kiperman S.L. Vvedenie v kinetiku geterogennyh kataliticheskih reakcij. M.: Nauka, 1964, p. 401.
286. Tyul'ga G.M., Gubanov V.A., Popova I.S., Petruhno L.A., Bolhovec B.M., Troichanskaya P.E., Dolgopol'skij I.M. Izuchenie reakcii ftorangidridov perftorkarbonovyh kislot s ftoridami shchelochnyh metallov. –ZhOrKh, 1973, t. 14, v. 11, p. 2339-2346.
287. Konshin A.I., Gubanov V.A. Ob ocenke ehffektivnosti sposoba ochistki hladonovyh rastvorov perftorpolioksametilenacetilftoridov. –Otchet predpriyatiya p/ya V-8415.
288. Patent SU 188404. Ftorsopolimer vinilidenftorida s 3,6,8,10,12-pentaoksaperftortridecenom dlya termomorozoagressivostojkih izdelij. / Grejs A.M., Ershov A.E., Zaharov V.Y. i dr.
289. Karcov S.V., Valov P.I., Sokolov L.F., Sokolov S.V. Rol' poverhnosti reakcionnogo sosuda v processe zhidkofaznogo okisleniya geksaftorpropilena. –Izv. AN SSSR, ser. khim., 1978, № 10. p. 2268-2272.
290. Patent SU 229185. Sposob polucheniya perftorpolioksametilenacetilftoridov. / Zaharov V.Y., Gol'dinov A.L., Borovnev L.M. i dr.
291. Borovnev L.M., Golubev A.N., Zaharov V.Y. i dr. Dinamika izmeneniya sostava gazovoj i zhidkoj faz v promyshlennom reaktore sinteza okisi geksaftorpropilena v hode operacii okisleniya. –Otchet predpriyatiya p/ya A-1619, 1983, inv. № 1089, 10p.
292. Borovnev L.M., Golubev A.N., Zaharov V.Y. i dr. Dinamika obrazovaniya perftorpolioksametilen acetilftoridov v processe zhidkofaznogo okisleniya geksaftorpropilena kislorodom na promyshlennoj ustanovke. –Otchet predpriyatiya p/ya A-1619, 1985, inv. № 1165, 22p.
293. Gol'dinov A.L., Borovnev L.M., Zaharov V.Y. i dr. Okislenie geksaftorpropilena molekulyarnym kislorodom v prisutstvii 1,2-dibromtetraftorehtana (hladona-114V2). Poluchenie okisi geksaftorpropilena. –Otchet predpriyatiya p/ya A-1619, 1982, inv. № 1072, 57p.
294. Patent SU 1128436. Plamyagasyashchaya smes'. / Borovnev L.M., Gusenkov M.V., Zhukova V.A. i dr.
295. Kravchenko N.N., Popov V.A., Ershov Y.A., Pomeranceva E.G. Nekotorye osobennosti fotoliza perftorirovannyh poliehfirov. –VMS, t. 23, № 3, 1981, p. 180-183.
296. Patent SU 3011822. Sposob regeneracii hladona-113. / Panitkova E.S., Berenblit V.V., Senyushov A.N. i dr.
297. Zejfman Y.V., Ter-Gabrielyan E.G., Ganbaryan N.P., Knunyantc I.L. Himiya perftorizobutilena. –Uspekhi khimii, 1984, t. 53, v. 3, p. 431-461.
298. Knunyantc I.L., Shokina V.V., Kuleshova A.D. Prisoedinenie galoidvodorodov k ftorolefinam. –Izv. AN SSSR, ser. khim., 1960, № 9, p.1693-1695.
299. Postovoi S.A., Mysov E.I., Zeifman Y.V., Knunyantc I.L. Nukleofil'naya kondensaciya oktaftorizobutilena s tetraftorehtilenom. –Izv. AN SSSR, ser. khim., 1982, № 7, p. 1586-1590.
300. Patent US 3389187. Dimer perftorizobutilena. / D.L. Miller. –Publ. 18.06.68. –RZh Khimiya, 1969, 17N3211.
301. Knunyantc I.L., German L.S., Dyatkin B.P. Reakcii ftorolefinov. SoobshchenieІІ. Vzaimodejstvie soedinenij ryada perftorizobutilena s aminami i ammiakom. –Izv. AN SSSR, otd.him.n., 1960, № 2, p. 221-230.
302. Patent JP 53-73504. Udalenie oktaftorizobutilena. / Kuroda Takehsi, Furukava Josiki, Macuoha Nu. –Publ. 30.06.78. RZh Khimiya, 1979, 10N2011.
303. Dorogimskij V.A., Kolomiec A.F., Sokol'skij G.A. O reakcii oktaftorizobutilena s ehtilenglikolem i glicerinom. –ZhOrKh, 1983, t.19, v. 8, p.1762-1763.
304. Patent US 129653. Sposob polucheniya geksaftorizomaslyanoj kisloty. / Knunyantc I.L., Cheburkov Y.A., Krasuskaya M.P. –RZh Khimiya, 1961, 9L75.
305. Borovnev L.M., Golubev A.N., Zaharov V.Y. i dr. Promyshlennye ispytaniya i vnedrenie sposoba ochistki gazov piroliza tetraftorehtilena ot perftorizobutilena. –Otchet predpriyatiya p/ya A-1619, 1981, inv. № 1048, 34p.
306. Gol'dinov A.L., Borovnev L.M., Zaharov V.Y. i dr. Uvelichenie stepeni ispol'zovaniya syr'ya v proizvodstve geksaftorpropilena putem bolee polnogo izvlecheniya oktaftorciklobutana iz kubovoj frakcii. –Otchet predpriyatiya p/ya A-1619, 1982, inv. № 1063, 16p.
307. Patent SU 180854. Sposob polucheniya geksaftorpropilena. / Borovnev L.M., Vinogradova A.P., Golubev A.N. i dr.
308. Borovnev L.M., Golubev A.N., Zaharov V.Y. i dr. Kompleksnaya pererabotka kubovoj frakcii proizvodstva tetraftorehtilena. –Otchet predpriyatiya p/ya A-1619, 1985, inv. № 15/107 DSP, 17p.
309. Patent SU 1336492. Sposob polucheniya 2-hlortetraftorehtilftorsul'fata. / Fokin A.V., Rapkin A.I., Tatarinov A.S. i dr.
310. Patent SU 1184808. Sposob polucheniya monoftorsul'fata broma. / Fokin A.V., Studnev Y.N., Rapkin A.I. i dr.
311. Patent SU 1239092. Sposob polucheniya tris(ftorsul'fata) broma. / Fokin A.V., Studnev Y.N., Rapkin A.I. i dr.
312. Patent SU 3982041. 3-(Poliftorpropionamido) propil-2-oksiehtildimetilammonij hloridy, obladayushchie gipotenzivnym dejstviem. / Fokin A.V., Kovalev G.V., Mazanov L.S. i dr.
313. Patent SU 3982040. N-Poliftoracil'nye proizvodnyeγ-amnomaslyanoj kisloty, obladayushchie gipertenzivnym dejstviem. / Fokin A.V., Kovalev G.V., Muhina N.V. i dr. Pol. resh. ot 26.09.86.
314. Patent SU 1334655. 3-(ω-Hlorperftoralkanoilamido)propil-dimetil-2-oksiehtilammmonij hloridy v kachestve poverhnostno-aktivnyh veshchestv. /Fokin A.V., Rapkin A.I., Studnev Y.N. i dr.
315. Patent SU 196359. Sposob polucheniyaω-hlorperftorkarbonovyh kislot i ehmul'gator. / Fokin A.V., Studnev Y.N., Rapkin A.I. i dr.
316. Patent DE 3034549. Sposob polucheniya perftorirovannyh ftorangidridov karbonovyh kislot. / Verner S. –Publ. 29.04.82, –RZh Khimiya, 1983, 17N5711.
317. Promyshlennye katalizatory i nositeli. Spravochnik. –Novosibirsk, 1972, p. 246.
318. T. Van der Plas. Tekstura i himiya poverhnosti uglerodnyh tel. V kn.: Stroenie i svojstva adsorbentov i katalizatorov. –M.: Mir, 1973, p. 436-441.
319. Patrik S. Uspekhi khimii ftora. –M., L., Khimiya, 1964, t.1, № 2, p. 336-379.
Recommended for publication by prof. S. M. Igoumnov
Fluorine Notes, 2014, 95, 1-2
