Received: May , 2014
Fluorine Notes, 2014, 94, 7-8
Synthesis of 1-iodo-3-perfluoroalkylpropanes and 1-iodo-4-perfluoroalkylbutanes
Bálint Menczinger, Gergely Jakab, Dénes Szabó and József Rábai*
Institute of Chemistry, Eötvös Loránd University, P. O. Box 32, H-1518, Budapest 112, Hungary
e-mail: rabai@elte.hu
Abstract: Simple method for the preparation of perfluoroalkyl-alkyl iodides is suggested.
Keywords: Fluorous building blocks, 3-perfluoroalkyl-propyl iodides, phosphorus(III) iodide.
3-(Perfluoroalkyl)-propyl iodides (CnF2n+1CH2CH2CH2I, 1a-e, n = 4, 6, 8, 9, 10) are important reagents and building blocks for fluorous chemistry [1], material science [2], polymer chemistry [3], and drug discovery [4].Their syntheses mostly based on the transformation of the precursor F-alcohols into iodides. For laboratory syntheses of such fluorous iodides using KI/phosphoric acid [5], NaI/chlorotrimethylsilane [4] or I2/P(C6H5)3/imidazole [6] reagent systems are well acceptable.
However, we demonstrate here that phosphorus (III) iodide - generated in situ from red phosphorus and iodine - reacts with 3-perfluoroalkylpropanols (2a-e) or fluorous-butanol (2f) to afford the title compounds in high yields and purities.

This process can easily be upscaled to the synthesis of several hundred gram quantities if the controlled introduction of iodine under an inert atmosphere is carried out by using a ‘hot-melt dropping funnel’ (Figure 1) [7].
The precursor 3-perfluoroalkylpropanols [8] can be made by the reduction of the corresponding F-iodohydrin adducts formed by the addition of perfluoroalkyl iodides to allylic alcohol in the presence of radical initiators, such as azobis(isobutyronitrile) [9] or the Cu(OAc)2/hydrazine hydrate redox system [10]. The latter F-iodohydrins are versatile intermediates allowing the preparation of valuable fluorous building blocks including fluorous allylic alcohols as well [11].
The products obtained were characterized by 1H-, 13C and 19F-NMR spectra and GC. The last traces of alcohol impurities can be removed by treatment of their ether solution with P2O5/SiO2 (SicaPent) [12], or by distillation over CaH2 [3].
Experimental
Perfluoroalkyl-propanols (2a-e) were prepared as reported in [8a], with GC purities >98%. Fluorous butanol 2f was prepared similarly but using homoallylic alcohol [13]. 1H-, 13C- and 19F-NMR spectra were recorded on a Bruker Avance 250 instrument using a 5 mm inverse 1H/13C/31P/19F probe head at room temperature. Chemical shifts (δ) are given in parts per million (ppm) units relatively to the internal standard TMS (δ =0.00 for 1H, δ =0.00 for 13C) and to CFCl3 as external standard (δ =0.00 for 19F). Melting points were determined on a Boetius micro-melting point apparatus and are uncorrected. The reactions were monitored by gas chromatography (Hewlett-Packard 5890 Series II, PONA [crosslinked methylsilicone gum] 50 m x 0.2mm x 0.5 mm column, H2 carrier gas, FID detection; Program: 120 °C, 5 min, 10 °C/min, 250 °C, 5 min, Inj.: 250°C, Det: 280°C).
Typical Procedure (TP):
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-Heptadecafluoro-11-iodo-undecane (1c)
A flame dried 1 liter volume four-necked glass reactor was equipped with a stirrer, thermometer, ’hot-melt dropping funnel’ and a condenser attached to an argon by-pass. Then the reactor was charged with perfluorooctylpropanol (2c, 574 g, 1.20 mol) and red phosphorus (13.0 g, 0.42 mol P), while the ’hot-melt dropping funnel’ with iodine (160 g; 0.63 mol I2). The mixture was heated with an oil-bath of about 100°C temperature until the fluorous alcohol melted, then stirring was started and the iodine addition was controlled by applying hot air flow with the aid of a heat gun; about 60 minutes required for completing iodine addition. The reaction mixture was heated and stirred for 2 h at 135-140°C internal temperature. Then the oil bath was removed and the reaction mixture was allowed to cool to ~40°C and the product was separated from the H3PO3 precipitate as a liquid and transferred into an Erlenmeyer flask. Partitioning between ether (1 L) and water (0.5 L) and decolorizing with aqueous NaHSO3 solution gave a colorless extract. It was dried (Na2SO4), filtered and the solvent evaporated by distillation. The pale yellow residue was purified by vacuum distillation to afford 607 g (86%) product, colorless liquid, bp = 111°C/16mmHg, which solidifies at room temperature and has an mp = 33-34°C (GC assay: 99.5%).
1H NMR (250 MHz, CDCl3) δ 3.24 (t, 2 H, CH2I),
2.08–2.33 (2 m, 4 H, C8F17CH2CH2);
19F NMR δ -81.9 (t, 3 F), -114.5 (2 F),
-122.7 (6 F), -123.5 (2 F), -124.2
(2 F), -127.0 (2 F); 13C NMR δ 32.30 (t, C8F17CH2);
24.66 (t, CH2CH2I); 3.65 (s, CH2CH2I).
This product displays equal 1H- 19F-, and 13C NMR spectra,
bp, mp and GC purity values that reported by our laboratory [12].
1,1,1,2,2,3,3,4,4-Nonafluoro-7-iodo-heptane (1a)
Rf4-propanol (2a, 33.4g, 120 mmol) was reacted with a slight excess of PI3 [prepared from red phosphorus (1.30 g, 42 mmol) and iodine (16 g, 63 mmol I2)] in a 250 mL volume heavy walled closed Pyrex tube for 3 h at 135-140°C and worked up to yield 34.0 g (73%) colorless oil, bp 70 °C/15 mmHg, (GC assay: 98%).
1H NMR (250 MHz, CDCl3) δ 3.23 (t, 2 H, CH2I),
2.06–2.32 (2 m, 4 H, C8F17CH2CH2);
19F NMR δ -82.3 (t, 3 F), -115.1 (2 F),
-125.5 (2 F), -127.2 (2 F); 13C
NMR δ 32.1 (t, C8F17CH2); 24.5 (t, CH2CH2I);
3.1 (s, CH2CH2I)
This product obtained showed agreeable physical properties and 1H- 19F-, and 13C NMR spectra to that reported in [3, 4].
1,1,2,2,3,3,4,5,5,6,6-Tridecafluoro-9-iodo-nonane (1b)
The Rf6-propanol (2b, 227g, 0.60 mol) was reacted with red phosphorus (6.50 g, 0.21 mol) and iodine (80 g, 0.315 mol I2) according to Typical Procedure for 2 h at 135-140°C and worked up to yield 237 g (81%) colorless oil, bp = 108°C/20 mmHg (GC assay: 99.5%).
1H NMR (250 MHz, CDCl3) δ 3.23 (t, 2 H, CH2I),
2.07–2.34 (2 m, 4 H, C8F17CH2CH2);
19F NMR δ -82.1 (t, 3 F), -114.8 (2 F),
-122.8 (2 F), -123.8 (2 F); -124.4
(2F); -127.2(2F). 13C NMR δ 32.2 (t, C8F17CH2);
24.6 (t, CH2CH2I); 3.0 (s, CH2CH2I)
This product showed agreeable physical properties and NMR spectra to that reported [2, 5].
1,1,1,2,3,3,4,4,5,5,6,6,7,7,8,8-Hexadecafluoro-11-iodo-2-trifluoromethyl-undecane (1d)
iso-Rf9-propanol (15.84 g, 30 mmol), red phosphorus (0.325 g, 10.5 mmol) and iodine (4.00g, 15.75 mmol I2) was reacted in a closed heavy walled Pyrex tube for 5 h at 135-140°C and worked up. Yield: 14.33 g (75%) colorless oil, bp = 110°C/1.0 mmHg (GC assay: 98% GC), mp = 28-29°C.
1H NMR (250 MHz, CDCl3) δ 3.25 (t, 2 H, CH2I),
2.07–2.35 (2 m, 4 H, C8F17CH2CH2);
19F NMR δ -72.2 - (-72.4) (m, 7 F),
-114.2 (2 F), -115.4 (2 F), -121.2 (2
F); -122.0 (4 F); -123.9 (2F). 13C NMR δ 32.3 (t, C8F17CH2);
24.7 (t, CH2CH2I); 4.1 (s, CH2CH2I)
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10-Heneicosafluoro-13-iodo-tridecane (1e)
Rf10-propanol (2e, 11.56g, 20 mmol), red phosphorus (0.22g, 7 mmol) and iodine (2.67g, 10.5 mmol I2) was reacted in a closed heavy walled Pyrex tube for 4 h at 135-140°C and worked up. The crude product (~14 g) was recrystallized from cyclohexane (90 mL) to afford 11.96 g (87%) white crystals, mp: 75-76 °C (GC assay 97%); Lit [14] mp = 80-82°C.
1H NMR (250 MHz, CDCl3) δ 3.25 (t, 2 H, CH2I),
2.08–2.34 (2 m, 4 H, C8F17CH2CH2);
19F NMR δ -81.2 (t, 3 F), -114.2 (2 F),
-122.2 (10 F), -123.2 (2 F); -123.9
(2F); -126.6 (2F). 13C NMR δ 32.3 (t, C8F17CH2);
24.6 (t, CH2CH2I); 3.8 (s, CH2CH2I)
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-Heptadecafluoro-12-iodo-dodecane (1f)
Rf8-butanol (10.1 g, 20 mmol), red phosphorus (0.22g, 7 mmol) and iodine (2.67 g, 10.5 mmol I2) was reacted in a closed heavy walled Pyrex tube for 9 h at 135-140°C and worked up. Yield 10.07 g (82%) colorless liquid, by short-path distillation (bath temperature: 135-140°C/1.0 mmHg), mp = 40-42°C (GC assay: 97%). Lit. [13] mp = 50-51°C.
1H NMR (250 MHz, CDCl3) δ 3.21 (t, 2 H, CH2I),
2.20–1.71 (m, 6 H); 19F NMR δ -81.4 (t, 3 F), -114.9 (2 F),
-122.5 (6 F),
-123.3 (2 F); -124.4 (2 F); -126.7 (2 F). 13C NMR δ 32.9 (s, C8F17CH2);
30.2 (t, one of CH2CH2CH2CF2 ); 21.7 (t, one of CH2CH2CH2CF2 ); 5.2 (s, CH2I).
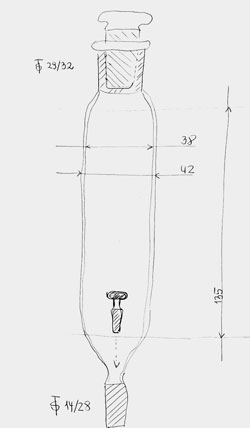
Figure 1. Hot-melt dropping funnel (cf. Ref. [7]).
The ‘hot-melt’ dropping funnel is an apparatus for the incremental addition of solids under an inert atmosphere. It was fabricated from an ST 29/32 female and an ST 14/28 male joint, both 60 mm long and a 120 mm length of 38 mm glass tubing. The above joints were flared to the glass tubing. Before using the apparatus it was fixed in an upright position and a small T shaped glass stopper (15 mm *6 mm *25 mm) was placed in to prevent the falling down of the solids when they are charged into the funnel. The ‘hot-melt’ dropping funnel can be charged with low melting solids such as iodohydrin or iodine. Finally the funnel was closed with an ST 29/32 stop-cock and fitted to the four-necked reactor. The addition rate of iodine was easily controlled by heating the lower end of the funnel by a ‘hot-air’ heat gun since the T-shaped glass stopper means no barrier for dropping down the melted iodine. The fabrication and use of an improved dropping funnel for liquid or melt additions has already been disclosed [15].
References
- (a) Vincent, J.-M.; Rabion, A.; Yachandra, V. K.; Fish, R. H. Angew.
Chem., Int. Ed. Engl. 1997, 36, 2346-2349;
(b) Gladysz, J. A.; Curran, D. P.; Horváth, I. T. (Eds.); Handbook of Fluorous Chemistry Wiley-VCH, Weinheim, (2004). - Tanaka, M.; Rastogi, A.; Toepperwein, G. N.; Riggleman, R. A.; Felix, N. M.; de Pablo, J. J.; Ober,C. K. Chem. Mater. 2009, 21, 3125–3135 3125.
- Sugiyama, K.; Hirao, A.; Nakahama, S.; Macromol. Chem. Phys. 1996, 197, 3149-3165.
- Suzuki, Y.; Kato, T.; Aoyama, H.; Misono, T.; Khlebnikov, V. A.; Mizutani, A.; Ohtake, Y.; Ikeda, T.; Takano, K.; Kwon, H.; Ho, P.-S.; Kim, J.-S.; Lee, W.-I.; Park, C.-H.; Lee, S.-H.; Ahn, S.-O.; (Chugai Seiyaku Kabushika Kaisha, Japan). PCT Int Appl. (2003), WO 2003004515 A1 20030116, 108 pp.
- Brace, N. O.; Marshall, L. W.; Pinson, C. J.; van Wingerden, G. J. Org. Chem. 1984, 49, 2361-2368.
- Kojima, M.; Nakamura, Y.; Ishikawa, T. Tetrahedron Letters 2006, 47, 6309–6314.
- Szijjartó, Cs.; Ivanko, P.; Takács, F. T.; Szabó, D.; Rábai, J.; J. Fluorine Chem. 2008, 129, 386–389.
- (a) Rábai, J.; Szíjjártó, Cs.; Ivanko, P.; Szabó, D. Synthesis
2007, 2581-2584.
(b) JSC "P&M-Invest", Synthesis of Organofluorine Compounds, Ed. Igoumnov S., Igoumnova E., Moscow, 2011, Vol. 2, Chapter 1, pp. 20-29, 31-32. - Brace, N. O.; J. Fluorine Chem. 1999, 93, 1-25.
- Igumnov, S. M.; Don, V. L.; Vyazkov, V. A.; Narinyan, K. E. Mendeleev Commun., 2006, 16 (3), p. 189-190.
- Ivanko, P.; Szíjjártó, Cs.; Szabó, D.; Rábai, J. Fluorine notes, 2012, Vol. 5(84).
- Rábai, J.; Abudurexiti, A.; Szabó, D. In: Gladysz, J. A.; Curran, D. P.; Horvath, I. T. (Eds.) Handbook of Fluorous Chemistry, Weinheim: Wiley - VCH, 2004. pp. 421-423.
- Alvey, L. J.; Meier, R.; Soós, T.; Bernatis, P.; Gladysz, J. A. Eur. J. Inorg. Chem. 2000, 1975-1983.
- Bayardon, J.; Sinou, D. J. Org. Chem. 2004, 69, 3121-3128.
- Mitchell, J., Jr., Henderson, J.W. Anal. Chem. 1950, 22, 374.
Recommended for publication by Prof. S.M. Igoumnov
Fluorine Notes, 2014, 94, 7-8
