Received: September, 2013
Fluorine Notes, 2014, 92, 1-2
The reactions of alkylation, arylation, alkenylation, and alkynylation of polyfluoroarenes and -hetarenes by their aromatic rings
T.D.Petrova, V.E.Platonov
N.N.Vorozhtsov Novosibirsk Institute of Organic Chemistry, Siberian Branch, Russian Academy of Sciences, pr. Akademika Lavrent,eva 9, Novosibirsk, 630090 Russia
e-mail: petrova@nioch.nsc.ru, platonov@nioch.nsc.ru
Abstract: Review considers the reactions of alkylation, arylation, alkenylation, and alkynylation of polyfluoroarenes and –hetarenes occurring in the aromatic ring and leading to the formation of CAr-C bond. Review contains information on the traditional methods of conducting such transformations which include, for example, reactions of polyfluoroarenes with electrophilic, nucleophilic, radical reagents. Along with these methods, reactions using of metal complex catalysis are considered. The application of metal complex catalysis expands the area the substrates used. This method includes cross-coupling reactions between metal or metal compounds and aryl halides catalyzed by transition metal complexes with various organic ligands. Kumada, Negishi, Suzuki, Stille, Hiyama, Heck, and Sonogashira reactions are presented. For polyfluoroarenes and –hetarenes CAr-Hal bond transformations involves also CAr-F bond.
Keywords: polyfluoroarenes and –hetarenes, alkylation, arylation, alkenylation, alkynylation, metal complex catalysis, palladium catalysts, cross-coupling reactions.
I Introduction
The formation of new C-C bonds is one of the most important chemical reactions of organic compounds since it allows the synthesis of a broad range of substances of different structure, whose properties may be of interest both alone and initial compounds or intermediates in the synthesis of practically important substances.
In a series of arenes, for reactions involving carbons that belong to aromatic ring, one of the ways to form CAr-C bonds are alkylation, arylation, alkenylation and alkynylation reactions.
Those transformations have long been known, and for many years their implementation made use of reactions with electrophilic, nucleophilic, and radical reagents, intramolecular cyclizations and rearrangements, transformations of preliminary introduced substituents, resulting in the formation of alkyl, aryl, alkenyl or alkynyl groups, as well as some coupling reactions catalyzed by metals (see, e.g., [1-3]. However, recently, along with those conventional methods, they started the development and intensive application of methods using catalysis by metal complexes having in mind to expand the area of those substrates use, to optimize the process conditions and product yields, and also for problem solving in cases when those conventional techniques wouldn’t give the desired results. Those methods include cross-coupling reactions between organometallic compounds and aryl halides catalyzed by transition metal complexes with various organic ligands, thus providing wide possibilities for the synthesis, and it appears currently to be among the most promising ways to C-C bond formation. The reaction of organomagnesium compounds is named Kumada reaction, that of organozinc compounds is Negishi reaction, that of organoborons is Suzuki reaction, that of organotins is Stille reaction, that of organosilicon compounds is Hiyama reaction, while the palladium-catalyzed introduction of alkenyl groups is known as Heck reaction, and that of alkynyl groups is called Sonogashira reaction. The patterns and mechanisms for those reactions are considered in a number of excellent reviews and monographs, e.g., [4-8]. For polyfluoroarenes and -hetarenes the above trends in the design of methods for CAr-C bond formation are the most evident, the only difference being that for those compounds their CAr-Hal bond transformation involves also CAr-F bond.
The data on the reactions of CAr-C bond formation in polyfluoroarenes, till 1995, were reviewed in 1997 by G. Brooke in his detailed review [9]. In this our review the published data on the reactions of alkylation, arylation, alkenylation, and alkynylation of polyfluoroarenes and -hetarenes are analysed over the last ~ 10 years.
To relevant polyfluoroarenes and –hetarenes here considered are compounds with at least two fluorines in the same aromatic ring, and their transformations include those of CAr-H and CAr-Hal bonds, here Hal = I, Br, Cl, F.
Our review summarises as well the studies on transformations of other substituents available in polyfluoroarenes or -hetarenes, but only if those substituents are directly linked to CAr, and those transformations result in the formation of alkyl or unsaturated groups. The haloalkylation reactions, including perfluoroalkylation, that had been reviewed elsewhere are not considered in this our article and only mentioned if they present some transformation of an intermediate on the way to the final product.
II Transformations involving CAr-H bond
1. Direct introduction of alkyl-, aryl- and unsaturated groups
1.1. Reactions in the presence of acid catalysts
1.2. Thermal and photochemical transformations
1.3. Reactions in the presence of bases
1.4. Metal complex catalysis
1.4.1. Use of palladium catalysts
1.4.2. Use of rhodium-based and nickel-based catalysts
2. Reactions with pre-activation of CAr-H bond by metalation
2.1. Formation of lithium derivatives and their reactions
2.2. Formation and transformation of organocopper derivatives
2.3. Formation and transformation of organoboron derivatives
2.3.1. Difluorophenylboronic acids
2.3.2. Trifluorophenylboronic acids
2.3.3. Pentafluorophenylboronic acid and polyfluorophenylborates
2.4. Formation and reactions of the polyfluorinated organozinc and organotin compounds
1. Direct introduction of alkyl-, aryl- and unsaturated groups
1.1. Reactions in the presence of acid catalysts
The polyfluoroarene inter- or intramolecular alkylation, classified with Friedel-Crafts reactions, may be exemplified by the obtaining of tris-(4-bromo-2,3,5,6-tetrafluorophenyl)methane via the reaction of 1-bromo-2,3,5,6-tetrafluorobenzene with CHCl3and AlCl3 (yield 22.4%) [10], chlormethylation of 1,3,5-trifluorobenzene with ClCH2OCH3in the presence of AlCl3in refluxing CS2(80%) [11], and also intramolecular cyclization of 3-chloro-N-(3',4'-difluorophenyl)-propionamide by AlCl3resulting in 6,7-difluoro-3,4-dihydro-1H-quinolin-2-one [12,13].
Methyl-3-(2',6'-difluorophenyl)-2-nitropropionate in surplus benzene solvent with excess of CF3SO3H readily undergoes phenylation at its 4' position resulting in a biphenyl derivative [14]. The tert-butylation of 3,5-difluorophenol at its 4 position proceeds with 54% yield if the substrate is heated (55°C) with tert-butyl methyl ether in the presence of zirconium tetrachloride [15].
The modification of conditions [16] did not give any positive result (yield 31%). The perfluorophenylation of perfluorobenzocycloalkenes (perfluorinated benzocyclobutene, indane, tetralin) and several derivatives thereof occur when those compounds interact with pentafluorobenzene and n-perfluoroalkyl tetrafluorobenzenes in SbF5medium (Scheme 1) [17-23].
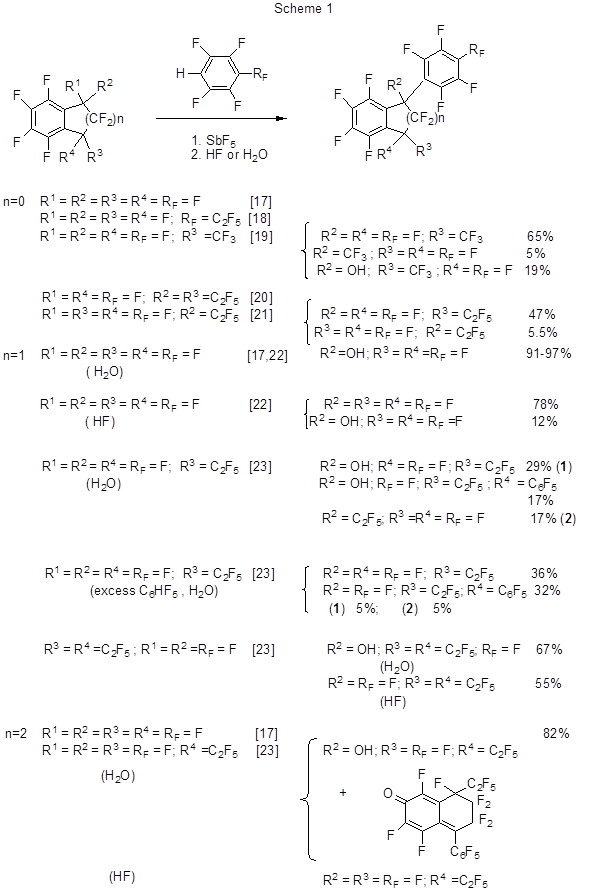
The reaction proceeds through the transformation of perfluorobenzocycloalkenes under the action of SbF5to give cationic intermediates that further interact with polyfluorobenzenes. The reactivity of those perfluorobenzocycloalkenes drops as the size of the alicyclic fragment increases [17]. As the polyfluorophenyl derivatives of perfluorobenzocycloalkenes thus formed in SbF5medium exist in the form of corresponding cationic salts only, the product separation from the reaction mixture is conducted by its treatment with HF. When treated with water it results in hydroxy derivatives formed through the interaction between water and the polyfluorinated cations. It should be noted that the reaction between perfluoro-1,1-diethylbenzocyclobutene and pentafluorobenzene, in the presence of equivalent amount of SbF5, results in perfluoro-1,1-diethyl-2-phenylbenzocyclobutene but its isolation fails because under the action of SbF5 and H2O it rapidly undergoes further transformation, so that the end products are mainly trienones (Scheme 2) [21].

Intramolecular Friedel-Crafts reactions resulting in the formation of heterocyclic ring are used in the obtaining of various polyfluorinated quinolinones starting from polyfluorinated anilides with a free ortho-position to amide group. Thus, heating of 3,4-difluoroanilide of β-chloropropionic acid with AlCl3 at 110°C [12] or 180°C [13] results in 6,7-difluoro-3,4-dihydro-1H-quinoline-2-one with yield 78% or 37% respectively (Scheme 3).
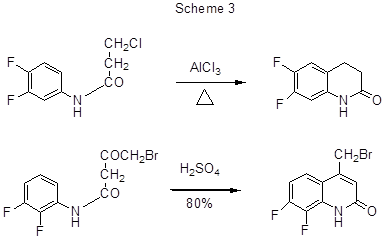
Scheme 3 shows another example of the anilide electrophilic cyclization [24].
Intramolecular alkylation comprising cyclization results from the interaction of polyfluorinated cinnamic acid anilides (di-, tri-and tetrafluoro- derivatives) with strong acids, in particular, CF3SO3H, or with AlCl3. It results in polyfluorinated quinolinones with sufficiently high yields (Scheme 4) [25-27]. The interaction with AlCl3, occurring under more rigid conditions, is followed by dehydrophenylation, and under similar conditions the same process takes place for non-fluorinated analogues as well.
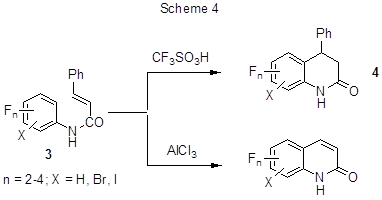
It is interesting that cyclization occurs despite the deactivating effect of several fluorine atoms. It is expected [25] that the transformation proceeds via an intermediate super-electrophilic dication (5) formed due to O, C-diprotonation (Scheme 5). The ease of cyclization is due to the substituents’ action both on the equilibrium concentration of dication 5, and on the energy of the cyclization transition state, which to some extent structurally similar to the intermediate (6).
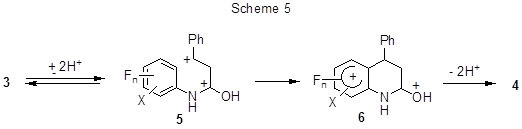
One-pot synthesis of polyfluorinated quinolines by the Skraup reaction was realised in the interaction between polyfluoroanilines, polyfluorohalogenated anilines and polyfluoroacetanilides with glycerine (Scheme 6).
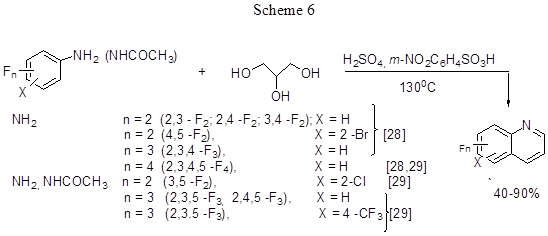
Fischer reaction between 2,5- difluorophenylhydrazine and disubstituted amide of 8-amino-1,4-dioxo-spiro[4,5]decane-8-carboxylic acid results in the derivatives of 5,8-difluoro-2,3,4,9 - tetrahydro-1H-carbazole (Scheme 7) [30].
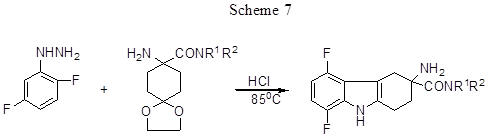
Aromatic amino-Claisen rearrangement in a series of N-(1-methyl-2-buten-1-yl) derivatives of fluorinated anilines in the presence of acidic catalysts leads to fluorinated ortho-alkylanilines (Scheme 8) [31,32].
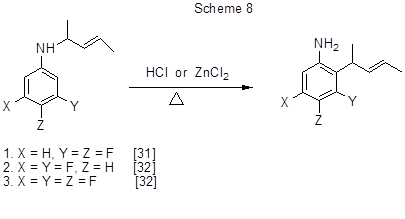
The reaction of N-alkyl-3,4-difluoroaniline [31] (case 1, scheme 8) was studied more explicitly. As a result, it was found that boron trifluoride etherate and trifluoroacetic acid may also be used as catalysts, and that besides of the isomer shown in Scheme 8 all catalysts favour the formation of another ortho-isomer with alkyl group at position 6 of the aromatic ring, and the alkylation product at position 6 of the original N-alkylaniline. The share of each of those three products varies depending on the catalyst, but in all cases the major compound is that with alkyl group at position 2. It is assumed that the rearrangement does not follow the concerted [3,3] sigmatropic mechanism, but the intermolecular mechanism comprising electrophilic catalytic cleavage of the N-C1bond and electrophilic attack by pent-3-en-2-yl cation of the ortho-position of the aromatic ring followed by deprotonation of σ-complexes. In the case of 2,4,5-trifluoro-N-(1-methyl-2-buten-1-yl)aniline the rearrangement does not occur [32], which is thought to be associated with the deactivation of position 6 by fluorine atom at position 5.
The amino-Claisen rearrangement takes place when the excess of 3,4-difluoroaniline reacts with 1-chloro-2-cyclopentene. Under the action of the original aniline hydrochloride the initially formed N-cyclopentenyl derivative is converted to a mixture of difluoroaniline ortho-cyclopentenyl derivatives, and 6-isomer dominates (Scheme 9) [33].

The reaction of this aniline and 1,4-benzoquinone with preliminary diazotization of difluoroaniline results in 90% yield to the product of difluorophenylation of 1,4-benzoquinone in position 2 [34].
1.2. Thermal and photochemical transformations
Thermal intramolecular Diels-Alder reaction of 2,4-difluorocinnamic acid ester containing a fragment of propargylic acid enables the synthesis of difluorodihydronaphthofuran derivatives (Scheme 10) [35].
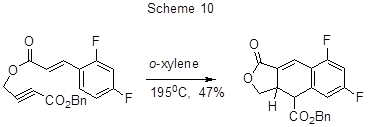
Thermal Claisen rearrangement of polyfluorinated phenols allylic esters in the presence of N,N-dimethylaniline makes it possible to produce polyfluorinated ortho-allylphenols (Scheme 11) [36-38].
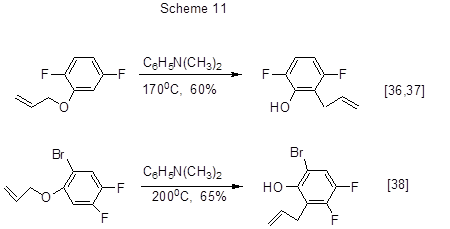
A convenient one-step method for the alkylation of polyfluorinated carbonyl derivatives of benzene or benzonitriles deactivated to Friedel-Crafts reactions is a photoinitiated radical chain reaction with alkylmercury halides [39]. Thus, oxidative tert-butylation of difluorosubstituted benzene derivatives is successfully carried out by the photolysis of their mixture with t-BuHgCl in the presence of a base, that is 1,4-diazabicyclo [2,2,2] octane (DABCO) used as a proton acceptor (Scheme 12).
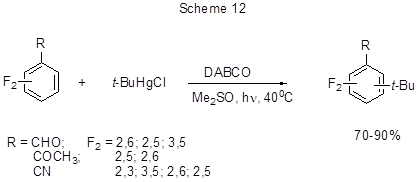
The mechanism of the process apparently assumes the participation of tert-butyl radical in the bimolecular aromatic homolytic substitution resulting in the formation of a radical adduct and further in that of a radical anion because the base (DABCO) eliminates proton from the radical adduct. Both the radical adduct and the radical anion are stabilised by the R substituent. The chain reaction is initiated by electron transfer from the radical anion to t-BuHgCl, regenerating tert-butyl radical (Scheme 13).
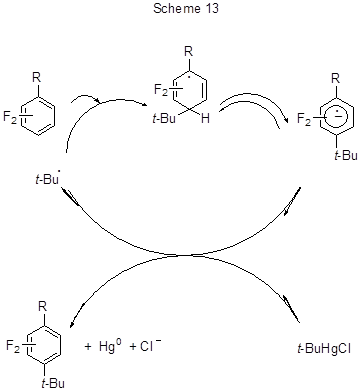
The reactions of tert-butylation take place predominantly in the para-position to R, and if it is occupied, the reaction does not occur. According to the Scheme 13 the regioselectivity must depend on the resonance stabilization of the radical adduct formed due to ortho- or para- attacks by the tert-butyl radical. However, the ortho-attack is less preferred because of the formation of an intermediate, this being a sterically strained radical with bulk tert-butyl group in its ortho-position. However, in the case of 2,4-difluorobenzonitrile, although the para-position is occupied, the reaction proceeds, and results in 46% yield of an ortho-tert-butyl derivative of difluorobenzonitrile. Therefore, ortho-substitution with respect to the nitrile group is activated, possibly due to the contribution of a linear resonance structure (Scheme 14), wherein the steric stress is less. In general difluoroacetophenones are less reactive than similar aldehydes or nitriles, perhaps due to sterically strained acyl group in the intermediate radical adduct.
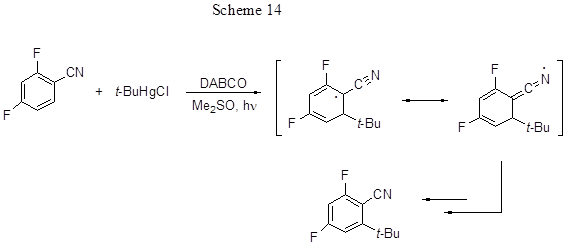
The method of oxidative coupling of 2-naphthol with 5,6,7,8-tetrafluoro-2-naphthol in the presence of a copper catalyst allows to obtain a moderate yield of tetrafluorobinaphthol (Scheme 15) [40].
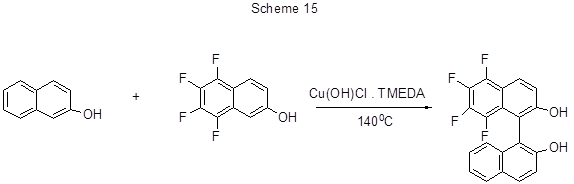
The reaction does not proceed with any other catalysts. The reaction temperature (at least 125°C) and catalyst (not more than 10%) are very important for this conversion. Under other conditions, binaphthol containing no fluorine atoms is formed.
1.3. Reactions in the presence of bases
Tert-butyl acetate group was introduced into position 4 of 2,6-difluoronitrobenzene by the reaction of 2,6-difluoronitrobenzene with tert-butylchloroacetate in the presence of potassium tert-butoxide (Scheme 16) [41).

Vicarious nucleophilic substitution in 2,6-difluoronitrobenzene with tert-butylethylmalonate and NaH in DMF followed by treatment with CF3COOH results in the ethyl ether of 2,3-difluoro-4-nitrophenylacetic acid [42].
1.4. Metal complex catalysis
The types of reactions mentioned in above chapters, particularly various modifications of Friedel-Crafts reactions, as well as other transformations, leading to the formation of new C-C bond, have been successfully used in organic chemistry for the synthesis of alkyl-, aryl-, alkenyl-, and alkynyl- derivatives of compounds belonging to different classes, including arenes and hetarenes, however, several limitations imposed on this type of reactions initiated research on new approaches to the preparation of these types of derivatives. During the recent decade the catalysis with complex catalysts based on transition metals it thought to be one of the most promising and successful approaches to the formation of C-C bond, thus opening wide synthetic possibilities. The use of catalysts for the transformation of inert C-H bonds is particularly important in the cases when the well-known methods are ineffective or wasteful. Transformations involving metal complex catalysts include either direct replacement of hydrogen at C-H bond by a desired group, or hydrogen pre-replacement by a metal to form an organometallic compound to react it further with a reagent containing the desired group. Usually those are cross-coupling reactions of arenes or metal-arenes with appropriate halogenated derivatives. In general, such transformations can be represented as follows:
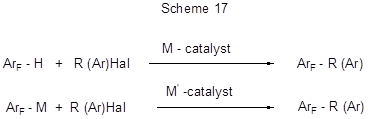
In what follows we shall consider the use of those approaches to a number of polyfluorinated arenes and hetarenes.
1.4.1. Use of palladium catalysts
This is a promising way for direct arylation of electron-deficient polyfluoroarenes as the usual reactions with electrophilic reagents are difficult for such compounds. Some researchers [43,44] have offered new conditions for direct arylation of penta-, tetra-, tri- or difluorobenzenes, 2,3,5,6-tetrafluoroanizol, 2,3,5,6-tetrafluorotoluene, and 4H-tetrafluoropyridine by the reaction of cross-coupling with a wide variety of aryl- and hetaryl- halogenides (Hal = I, Br, Cl) in the presence of a base (K2CO3, 1.1 equiv.) and palladium catalyst Pd(OAc)2 (1-5 mol%) and P(t-Bu)2Me - HBF4 (2-10 mol%), or formed in situ from Pd(OAc)2 and 2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl (S-Phos) (Scheme 18).
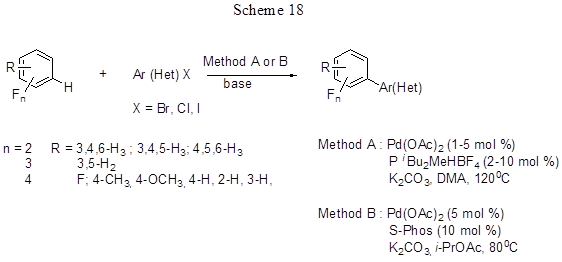
The reaction with pentafluorobenzene proceeds at 80°C in isopropyl acetate, those with other polyfluoroarenes proceed in DMA at 120°C. Earlier successful arylation of pentafluorobenzene was carried out by cross-coupling of pentafluorophenyl boron derivatives with aryl iodides (for more information on this type of reactions, see 2.3.). In the present case there is no need to obtain the precursor pentafluorophenylboronic derivative, it simplifies the process and, of course, refers to the advantages of this method. When polyfluoroarenes interact with chlorobromobenzene the reaction involves C-Br bond. In the case of dibromoarenes double arylation is observed. Brominated arene derivatives that involve various substituents either in one or in both ortho-positions react very well. Chlorinated arenes both activated by para-methyl group or those deactivated react actively. In the case of iododerivatives the best results are obtained with small addition of Ag2CO3[44], that helps to remove the formed iodide salts inhibiting the process. Tri- and di-fluorobenzenes react with lower yields than tetra- and penta- fluorobenzenes (for pentafluorobenzene the yield is 65-98%, while for difluorobenzenes the yields are 29-42%), i.e. for polyfluorinated arenes there is obvious inversion of reactivity as to compare with conventional aromatic electrophilic substitution, because more acidic C-H bonds in arene react preferentially. If a fluoroarene contains a number of hydrogen atoms, either diarylation or triarilation may take place resulting in a mixture of products. For 1,3-difluorobenzene the reaction involves the most acidic proton in position 2 (product yield 85%). The study on competitive reactions showed that the reactivity of C-H bond in fluoroarene is parallel to its acidity, and in the presence of two C-H bonds the reaction involves the more acidic one that is ortho-positioned to fluorine atom. The possible reaction routs are exemplified in Scheme 19 by the arylation of pentafluorobenzene with aryl bromide. According to the calculated results [43], the reaction does not include the formation of metalcyclic intermediates. The key step is the cleavage of C-H bond that includes the concerted metalation of fluoroarene and H transfer either to the catalyst carbonate ligand (path C2) or to the Br ligand (path C1). The metalation sensitivity immediately depends on the acidity of the ruptured C-H bond. Path C2 has got the lowest energy barrier and it better fits the available experimental data, so it is assumed to be the most probable path, though path C1 also cannot be completely rejected and it is possibly realized in some cases.
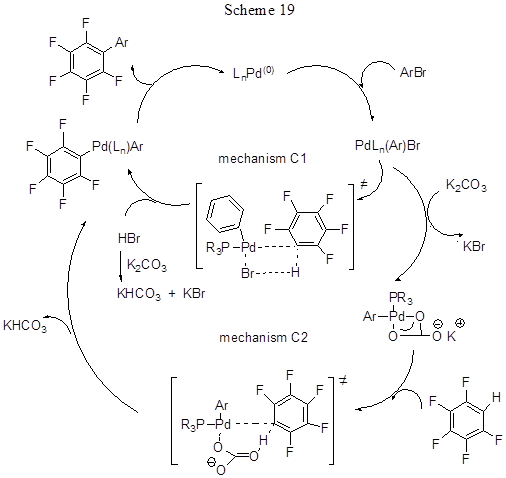
The considered reactions are scalable, and if using approximately equimolar amounts of two arylic components they result in polyfluorinated biphenyl products that may be important for medical chemistry, electron transport devices, light guide organic diodes, and elements in design of the liquid crystal [43]. Similar conditions for pentafluorobenzene arylation resulting in pentafluorobiphenyl derivatives are applied in the reaction with the bromophenyl fragment of a series of C-2'-deoxyribonucleotides (yield 64% [45]) and 3-(trifluoromethylacetoxy)-4-methoxybromobenzene (53% yield [46]).
An interesting variant of direct pentafluorobenzene arylation by p-bromotoluene using Pd-catalyst was proposed in [47]. Similar to the above mentioned studies, the reaction was carried out in the presence of a base (in this case, K3PO4) at 120° C (DMF solvent) and using Pd(OAc)2catalyst that does not contain bulky electron-donating phosphines for ligands, preferably from the standpoint of economy and production, but pivalic acid was added as a co-catalyst. Carboxylate anion contributes to lowering of the C-H bonds cleavage energy and may serve as a proton-carrier from substrate to inorganic base; and together catalyst plus co-catalyst provide the deprotonation-metalation process, according e.g. Scheme 20 presenting the mechanism of xanthine arylation under the action of n-bromotoluene in the presence of Pd (OAc)2and pivalic acid. The yield of the product arylation of pentafluorobenzene by this way is 80%.

Oxidative coupling of 1,4-difluorobenzene with 1,2-naphthoquinone in the presence of Pd(OAc)2, and CH3COOH, and following the technique [48] described for 1,4-dichlorobenzene resulted in the formation of 4-(2,5-difluorophenyl)-1,2- naphthoquinone (Scheme 21) [49].

Activation of C-H bond in 3,4-difluoroacetanilide by palladium complex occurs in the Mizoroki-Heck oxidative reaction between the anilide and butyl acrylate [50]. The main direction of the reaction is alkenylation to position 6 (product yield 60%). In the presence of the trimethylsilyl group at position 2 a 2-alkenyl derivative is formed (85%) (Scheme 22).
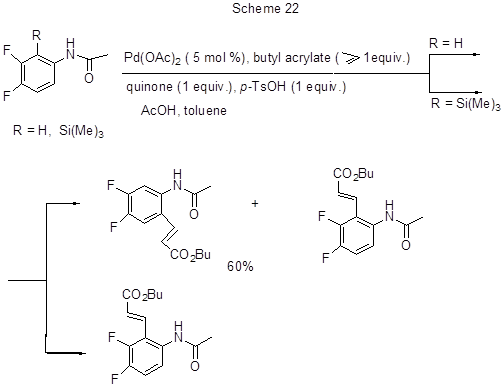
The reaction proceeds via the formation of cyclic palladium complexes due to the substitution of corresponding ortho-hydrogen, or silicon atoms with palladium. Those complexes were isolated and characterized by spectroscopic and X-ray diffraction methods. Those transformations require the presence of acetamide group, because it facilitates linking of electrophilic palladium to the ring, and favours the formation of a new C-C bond in its ortho-position. Indeed, the reaction does not occur with free amines.
The interaction between dihalogenated anilines and dihalogenated pyridines in the presence of palladium catalyst and a base in a one-pot process leads to α-carbolines. The reaction proceeds in two steps via palladium-catalyzed arylamination followed by intramolecular arylation. In this manner 5,8-difluoro- α-carboline was produced (Scheme 23) [51].
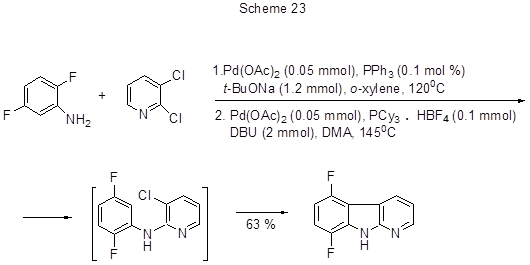
The best are conditions, when at the first process step triphenylphosphine is a ligand of the palladium catalyst, and tricyclohexylphosphine complex with HBF4, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) are further added, and the reaction temperature rises.
The palladium-catalyzed exclusively 5-exo-dig hydroarylation with intramolecular cyclization resulting in some derivatives of 9-benzylidene-9H-fluorene occurs in the case of ortho-alkynylbiaryls (Scheme 24) [52].
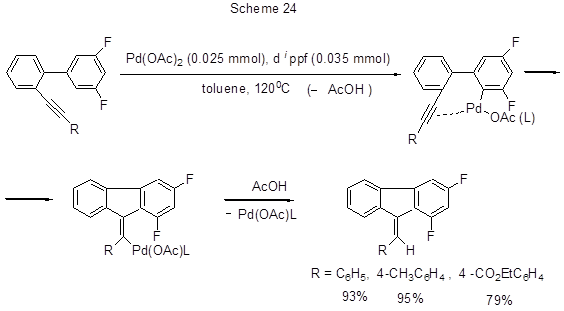
The reaction proceeds due to the activation of CAr-H bond thanks to ortho-palladation, and insertion of palladium into triple bond to produce a vinylpalladium intermediate which converts to the finish compound via protodepalladation, the catalyst being regenerated.
Another convenient variant of the palladium-catalyzed method for direct arylation of electron-deficient polyfluoroarenes is their cross-coupling with arylboronic acids in the presence of an oxidant (Scheme 25) [53].
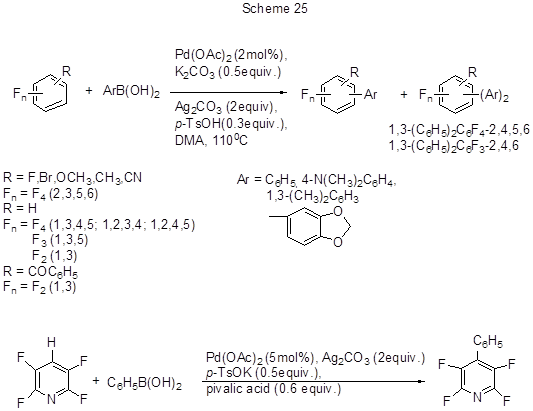
In reactions with pentafluorobenzene, along with said aryl boronic acids, some other acids with Ar = 4-RC6H4(R = Cl, CH3, OCH3, CHO, N(CH3)2) were applied. The reaction success is due to simultaneous adding to Pd (OAc)2and oxidant both of a weak base that promotes cleavage of the acidic C-H bond, and of a weak acid, that slows down transmetalation of arylboronic acids, that otherwise would lead to their unwanted homocoupling. Using the reaction of pentafluorobenzene (0.2 mmol) with phenylboronic acid as an example, it can be shown that the best conditions for the formation of pentafluorobiphenyl (yield 90%) assume the use of 2 mol% Pd(OAc)2, 0.5 eq. K2CO3, 0.3 eq. p-toluic acid and 1.2 equiv. phenylboronic acid. The effective oxidant is Ag2CO3(2 eq.), reaction temperature is 110°C, the solvent is DMA. The presence of donating substituents in aryl boronic acid molecule favours biaryl formation; however, the reaction does not occur with ortho-methyl-substituted phenyl boronic acid. The lower reactivity of aryl boronic acids with electron-withdrawing substituents might be offset by the replacement of base (e.g., using KF instead of K2CO3) and increase it in amount. The reactivity of polyfluoroarenes directly depends on the acidity of C-H bond, and it dictates the choice of the base. For the transformation of more acidic C-H bonds in 2,3,5,6-tetrafluoropyridine and 2,3,5,6-tetrafluorobenzonitrile they use 0.5 eq. K-salt of n-toluic acid as a base and 0.6 eq. pivalic acid, the ratio polyfluoroarene-to-arylboronic acid being 3:1. For polyfluoroarenes with less acidic C-H bonds (isomeric tetrafluorobenzenes, bromotetrafluorobenzene, 1,3,5-trifluorobenzene, 1,3-difluorobenzene, and 2,4-difluorobenzophenone) a combination of 0.5 equiv. K3PO4 and 0.6 eq. pivalic acid is used. Although some polyfluoroarene molecules have multiple positions for arylation, their monoarylation is mainly realized by the most acidic C-H bond, e.g., that located between two fluorine atoms. Thus, 1,3-difluorobenzene arylation occurs at position 2. In the case of 1,3,5- trifluorobenzene, a mixture of mono-and diaryl derivatives is produced in the ratio 4.5: 1, while for 1,3,4,5-tetrafluorobenzene it is a mixture of mono- and di- derivatives with ratio 4.6:1. The arylation of 2,4-difluorobenzophenone occurs exclusively at the meta-position to its benzoyl group and at the para-position to fluorine .
The authors propose the following scheme for the transformations, starting with the formation of an arylpalladium intermediate through B→Pd trans-metallation followed by palladation of fluoroarene by concerted metalation - deprotonation and reductive elimination of Pd (0) (Scheme 26).
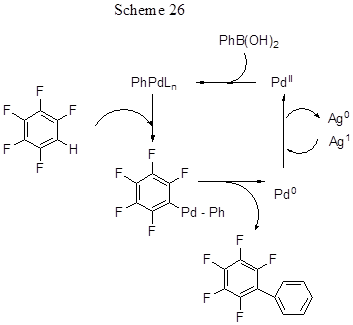
1.4.2. Use of rhodium-based and nickel-based catalysts
Polyfluoroaryl boronic acid derivatives may also be used as arylating agents, e.g. in the case of quinones. Thus the reaction of trifluoro-(3,5-difluorophenyl) borate with 1.5 molar excess of naphthoquinone and catalytic amount rhodium catalyst results in the formation of 2-difluorophenyl derivative of naphthoquinone, but the compound yield is not high (22%, Scheme 27) [54]. A by-product of the process is 1,4-hydroxy-naphthalene.

In the presence of rhodium catalyst the reaction of 3,4-difluorophenylboronic acid with 1-(1-benzyl-2,5-dihydro-1H-pyrrol-3-yl)ethanone occurs through the addition of fluorinated aryl fragment to the double bond of 2,5- dihydropyrrol ring (Scheme 28) [ 55].
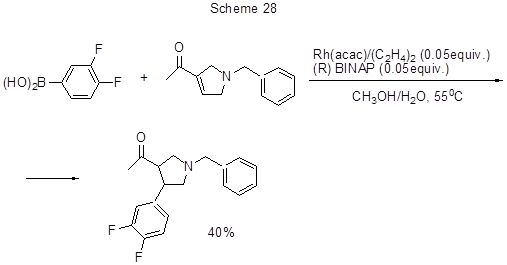
Amino-Claisen rearrangement of N-1-propargylanilines with acceptor substituents in the ring including fluorine atoms occurs at heating in hexafluoroisopropyl alcohol medium at the presence of rhodium catalyst and results in 2- or 2,3-substituted indoles [56]. This approach was used for the synthesis of 2,3-dimethyl-4,6- difluoroindole (Scheme 29). It is assumed that the reaction proceeds via intermediate ortho-allenylaniline derivative, due to amino-Claisen rearrangement, that further converts to an indole derivative via intramolecular cyclisation.

The real process catalyst is an alkoxy adduct of RhH(CO)(Ph3P)3with solvent; the adduct was isolated and characterized (Scheme 30).

Efficient alkenylation and alkynylation of polyfluoroarenes via activation of their CAr-H, favoured over that of CAr-F, occur in their interaction with 1,3-dienes, vinylarenes, and alkynes in the presence of a complex nickel-catalyst [57]. The method advantage is that it does not require pre-deprotonation of acidic hydrogen of polyfluororarene by the action of organometallic base, taken in stoichiometric amount, followed by a reaction with an electrophile. In this case to activate CAr-H bond it is sufficient to use some nickel catalyst in catalytic amounts (5-10 mol%). It is interesting, that although usually for Ni(0) complexes, oxidative insertion at C-F is favoured over that at C-H; in this case C-H bond participates in the reaction (Scheme 31).
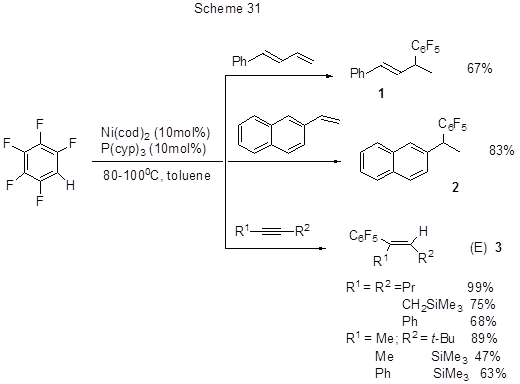
Sterically strained non-symmetric alkynes are stereoselectively added resulting in cis-adducts with bulky substituents exclusively at trans-positions to the polyfluorinated ring. For 4-octyne its reactions were studied with 2,3,5,6-tetra- and 3,5-difluoropyridine, 1,2,3,4- , 1,2,3,5- and 1,2,4,5-tetrafluorobenzene, 1,2,3- trifluorobenzene, and 1,2- and 1,3- difluorobenzene, 2,3,5,6-tetrafluoroanizole and methyl ester of 3,5-difluoro-benzoic acid. If polyfluoroarene molecule contains more than one hydrogen atom the reaction involves the second CAr-H bond to form dialkenyl polyfluoroarenes; the ratio of mono- to di-products depends on the reactants ratio: excess of 4-octyne favours disubstitution, while the excess of polyfluoroarene favours mono- alkenylation. Increasing the number of fluorine atoms facilitates the reaction. The most activated C-H bonds are those at ortho-positions to fluorine.
The reaction with 1-phenyl-1,3-butadiene occurs at the terminal double bond (Scheme 31). The reaction with 2-vinylnaphthalene (Scheme 31) results in 1,1-diarylethane. The reaction mechanism is exemplified by that of pentafluorobenzene represented in Scheme 32.
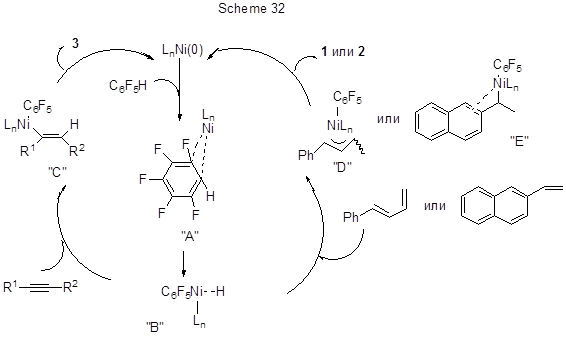
The reaction begins with the formation of η2-arene complex "A" followed by oxidative addition of Ni to the arene C-H bond and the formation of "B". The coordination is preceded by introduction of alkyne in Ni-H bond. The coordination can avoid steric repulsion between the most bulky R2group and C6F5-ring at Ni-centre. The reductive elimination results in alkenylated polyfluoroarene (3) and regenerates Ni (0) compound. With alkenes π-allyl complex "D" or benzylnickel complex "E" are formed respectively, their reductive elimination results in products (1), (2), and the regenerated Ni (0).
2. Reactions with pre-activation of CAr-H bond by metalation
2.1. Formation of lithium derivatives and their reactions
The well- known and widely used method for alkylation or arylation of aromatic molecules is replacing of aromatic hydrogen with lithium followed by reacting of the formed lithium-organic compound with an electrophile. The presence of fluorine atoms in polyfluoroarene molecule makes adjacent protons more acidic thus facilitating their interaction with lithium bases. The most acidic proton at the ortho-position to fluorine undergoes lithiation. In the case of two fluorine atoms at meta-positions CAr-H bond between them is subjected to lithiation. Lithiation occurs at low temperature and it is generally conducted in the presence of N, N-diisopropylamine or N,N,N',N'-tetramethyl-1,2-diaminoethane (TMEDA). For the reactions with methyl iodide it is the rout for methylation of 2,4,5-trifluorobenzoic acid to 2,4,5-trifluoro-3,6- dimethylbenzoic acid, and 3-chloro-2,4,5-trifluorobenzoic acid to 3-chloro-2,4,5-trifluoro-6-methylbenzoic acid [58, 59],tert-butyl(2,3-difluorobenzyloxy)-dimethylsilane to 4-methyl derivative [60], 2,4-difluorobromobenzene to 3-methyl-2,4-difluorobromobenzene [61], 3,5-difluorobromobenzene to 4-methyl-3,5- difluorobromobenzene [ 62], 1-cyclopropyl-6,7-difluoro-2-methoxy-1H-quinazoline-2,4-dione to 5-methyl derivative [63]. The reaction of methyl iodide with 1,3-difluoro-5-(3',5'-dimethoxybenzyloxy)benzene results in 90% yield of 2-methyl derivative, which is methylated to 2,4-di-and 2,4,6-trimethyl derivative in similar manner (Scheme 33) [ 64].
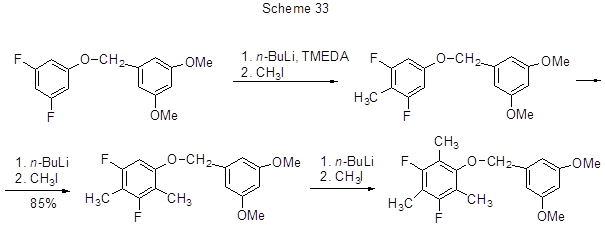
In this case, the position for lithiation is determined not only by ortho-orienting effect of fluorine, but also by oxygen-lithium reagent complexation, however, the first effect dominates and governs the location of the methyl group first entry (position 2). This is also favoured by metha-positions of fluorines and oxygens. For compounds with other positions of both, their benzylic protons are selectively lithiated, albeit with low yields.
Other examples of monomethylation or dimethylation of polyfluorinated fragments with preliminary lithiation and treatment with methyl iodide are described in [65, 66], and those with dimethylsulphate in [67]. 2,3-Difluoroalkylbenzenes (Alk = n-Bu,i-Bu, Et, n-Pr, cyclohexylmethyl) are produced from o-difluorobenzene in the reaction with n-butyl lithium and appropriate alkyl iodide in the presence of TMEDA [68]. The lithiation of 3,4-difluorobromobenzene at position 2 and subsequent reaction with a cyclopentene triflate results with good yield in a product containing a benzyl C- C bond (Scheme 34) [69].
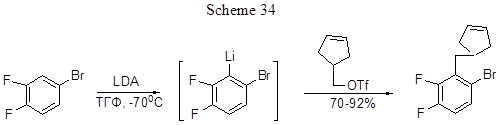
Non-catalyzed anionic interaction of aryllithium derivatives with 1,2-dibromobenzene provides direct access to ortho-,ortho-halogenated biaryls, which may be of practical importance because biaryl moiety makes part of many biologically active compounds used in pharmaceutical and liquid crystalline structures. This transformation is realized by reacting m-difluorobenzene [70], or some its derivatives [71] with 1,2-dibromobenzene (Scheme 35).
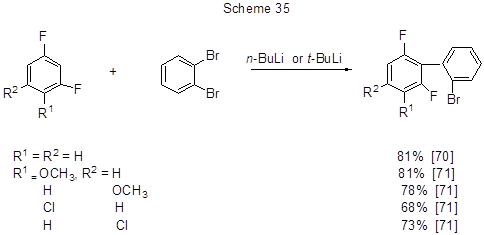
The transformation mechanism involves the formation of aryl lithium intermediates through the Hal→M and H→M exchange, and the participation of highly reactive dehydrobenzene, that is produced from o-dibromobenzene via the conversion of unstable lithium derivatives. It is followed by nucleophilic addition of a rather stable polyfluorinated lithium derivative to dehydrobenzene, and this is the rate-determining step. The next step is bromine transfer from the original o-dibromobenzene, resulting in the final ortho polyhalogenated substituted biaryl (Scheme 36). The advantage of the method is that it does not require complicated catalysts for coupling .

2-Bromo-2'6'- difluorobiphenyl is produced from 1,3-difluorobenzene and -bromochlorobenzene in similar manner (yield 66%) [72].
It is assumed that the intermediate dehydrobenzene is also produced through the lithiation of 2,6-difluoro-trimethylsilylbenzene. The said dehydrobenzene reacts with lithium predecessors resulting in isomeric trifluorobiphenyls (Scheme 37) [73].
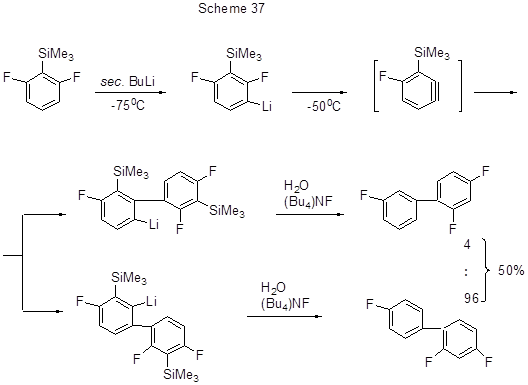
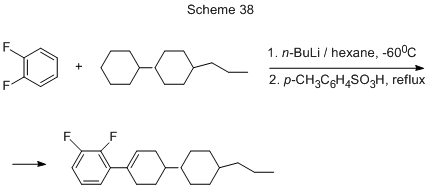
The cycloalkylation of o-difluorobenzene under the action of 4-(trans-4-propylcyclohexyl)cyclohexane with simultaneous dehydrogenation is realized as well, its final product being 2,3-difluoro-1-[trans-4-(trans-4-propylcyclohexyl)cyclohexenyl]benzene (yield 48 %, Scheme 38) [ 74].
Organolithium compounds formed from polyfluoroarenes may add to carbonyl groups of aldehydes or ketones resulting in polyfluoroarene oxyalkylation products. Some examples of such conversions are shown in Scheme 39.
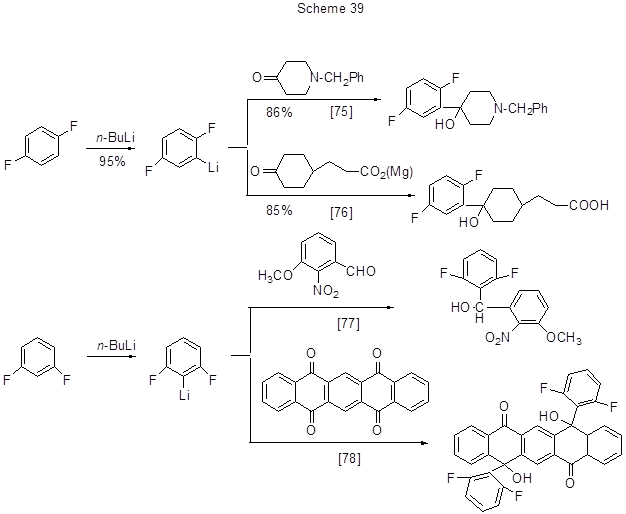
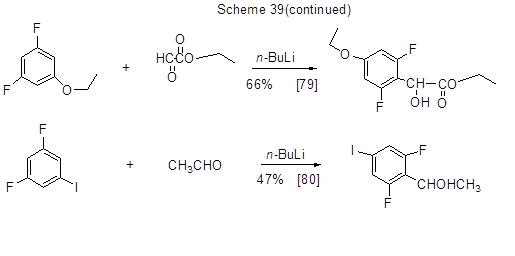
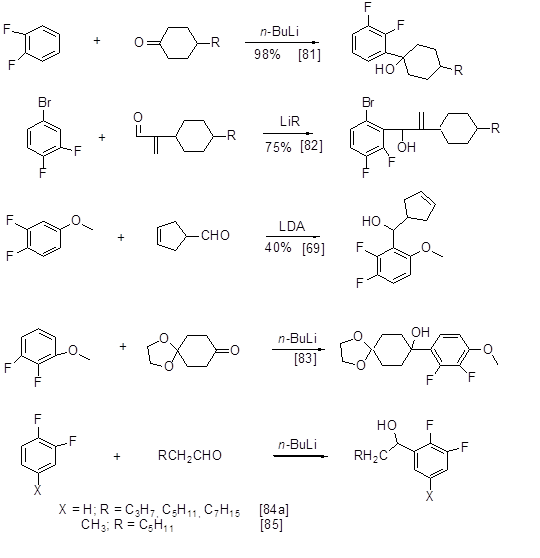
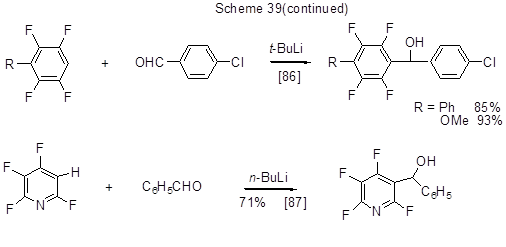
Further to the said transformations of polyfluorinated lithiumarenes, some of them are used for feedstock in organometallic synthesis of other organometallic derivatives, inter alia, compounds of copper, boron, etc., via transmetalation process.
2.2. Formation and transformation of organocopper derivatives
A general method for copper-catalyzed arylation, alkenylation, and benzylation of Csp2-H bond, designed for a wide range of compounds with a pKa less than 35, is successfully applied to polyfluoroarenes. It comprises copper salts-catalyzed reacting of arenes containing two or more fluorine atoms in their rings, with the corresponding halogen derivatives in the presence of some bases [88-91]. Both iodides and bromides, and occasionally, chlorides, e.g., heterocyclic ones, that may comprise donor or acceptor groups, are applicable. Iodides are preferable in the case of less reactive species (e.g., mesitylene derivative). The best results are obtained when CuI catalyst is combined with phenanthroline ligands. K3PO4 is the most acceptable base, but for difluorosubstituted substrates the best results are those with lithium alkoxides. DMF or DMF/xylene is used as solvent. The method provides a convenient and cheaper alternative to other metal catalysis, e.g. palladium, ruthenium or rhodium catalysts (Scheme 40).

The reaction proceeds most efficiently at the most acidic C-H bond located between two fluorines. In the case of 1,2,3,4-tetrafluorobenzene, where such bonds are not available, the reaction with p-iodotoluene proceeds with only 10% yield [89]. If a molecule contains two halogens, be they similar [89, 90] or different [91], both of them participate in the reaction and therefore two aryl groups Ar are introduced into the molecule. The possible reaction mechanism is shown below (Scheme 41).
Scheme 41
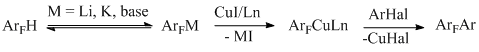
It is assumed that under action of a base the of acidic bond CAr-H undergoes deprotonation, and an ArFM compound thus formed is further transmetallated to an organocopper compound, which being added to ArHal, results in the final product. In support of this scheme an intermediate complex C6F5Cu-phenanthroline is produced, isolated and characterized in the reaction with pentafluorobenzene, the complex reacts with aryl iodide to give biaryl [89].
It is found that a direct catalysed reaction of polyfluorinated arene coupling with compounds containing no halogen atoms is possible. Therefore, the reaction between polyfluorene and terminal alkynes in the presence of a copper catalyst in air results in polyfluoroarylalkynyls with yield of 25 to 76% (Scheme 42) [ 92].
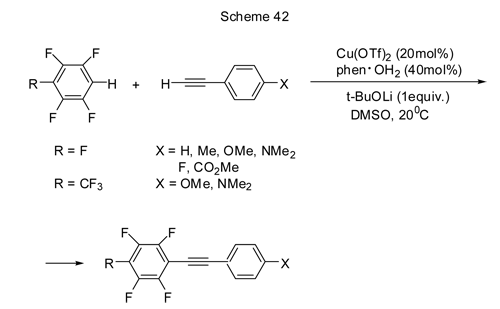
The presence of donor substituents X favours the yield of the coupling product. With R=CF3 even with donor X substituents the yields are lower than those with R=F. Tri- or difluorenes do not enter the process, may be because of insufficient acidity of C-H bonds in those compounds. Alkylalkynes do not react at those conditions as well.
2.3. Formation and transformation of polyfluoroarylorganoboronic derivatives
Arylboronic compounds, mostly arylboronic acid or its derivatives produced from corresponding aryl lithium derivatives and boron compounds, undergo cross-coupling with aryl halides, triflates or tosylates, in the presence of a base, catalyzed by transition metal complexes, most often those of palladium or nickel. Thus there is a splitting off halogens or OSO2R-groups, and new C-C bonds are formed due to combination of aryl-aryl fragments. This transformation, named Suzuki reaction, opens numerous synthetic possibilities being one of the most promising methods for the manufacture of bi- and polyaryls, many of which have practically useful properties or serve as a basis for the preparation of compounds having such properties [93]. In general, the reaction can be represented as follows (Scheme 43) [5, p.25, 26].

Chlorides are least active in this reaction due to the higher dissociation energy of C-Cl bond, resulting in lower ability of this bond to add oxidatively Pd (0) or Ni (0), while this is a key step of the process. In Suzuki reaction, as in all transformations with metal complex catalysts, the important role belongs to the choice of base and catalyst ligands, and the amount of catalyst used. This reaction, with all its features, advantages, disadvantages and mechanism details is discussed comprehensively in a number of reviews, e.g., [4, 94], and monographs [5-8]. In this chapter we analyze its application to a number of polyfluoroarenes, while among the considered polyfluoroboroarenes there are compounds which lithium precursors are produced when lithium substitutes either proton at CAr-H bond, or halogen at CAr-Hal bond. For more details of CAr-Hal → CAr-Li transformations, see III.
2.3.1 Difluorophenylboronic acids
2,4-Difluorophenylboronic acid (1) and its 3,4- (2), 3,5- (3) and 2,5- (4) difluoroisomers react with 3-bromophenylmethylacetate, in the presence of NaOH, and a triphenylphosphine derivative of palladium used for a catalyst, and the process involves the substitution of bromine [95]. Similarly, reaction 1 with 4-bromobenzaldehyde occurs using the same catalyst, and K2CO3as base (Scheme 44) [96].
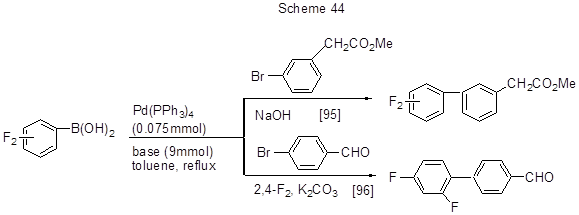
Through the interaction of 1 with methyl-2{[(trifluoromethyl)sulfonyl]oxy}benzoate methyl-2',4'- difluoro-2-carboxylate is produced (Scheme 45) [ 97].
Suzuki reaction between acid 1 and 4-bromoacetophenone conducted in the presence of K2CO3and various palladium complex catalysts results in high yields of the cross-coupling products [98, 99].
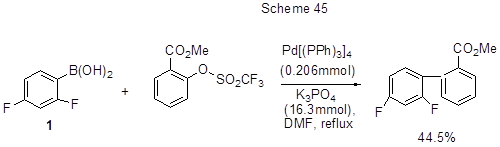
It is found that when the ligand chosen for PdCl2is electron-donating and sterically bulky dibenzyl -N-(diisopropyl)phosphoramidite P(OBn)2N(i-Pr)2, while the base is Na2CO3, it is possible to use only 0.01 mol % of the catalyst, to carry out the reaction at room temperature, and to prepare the product with a yield of 92% [100].

The reactions of compound 1 with halogenated heterocycles containing a halogen atom directly on the heterocycle or in the heterocyclic aryl component are exemplified in Scheme 46 (asterisk denotes the substitutable halogen atom). In most cases palladium catalysts, are Pd(PPh3)4or PdCl2(PPh3)2, while K2CO3, Na2CO3, Cs2CO3are used as bases.
For more examples of such reactions between compound 1 and various heterocyclic halides see [107-115]. The process using microwave radiation is disclosed in [116]. Generally, any such reaction constitutes a step of a multistep process for the production of complex substances with promising properties.
Suzuki reaction with 3,5-difluorophenylboronic acid 3 is illustrated by Scheme 47.

3,5-Difluorodiphenylmethanes are produced with high yields (70-97%) by reacting compound 3 with benzyl chloride or bromide and using Pd[(N-succ.)Br(PPh3)2] for the catalyst [121]. Polyfluorinated alkylcyclohexyl derivatives of bi- or terphenyls are obtained by Suzuki cross-coupling reactions between acid 3 and 4- or 4'- alkylcyclohexyl derivatives of 1-iodo-2-fluorobenzene or 3-fluoro-4-iodobiphenyl respectively in the presence of Pd/C and K2CO3(yields from 65 to 87%) [122, 123]. An example of triflate group substitution with 1,3-difluorophenyl group in a cyclohexene derivative by its reaction with 3 is shown in [124] (55% yield).
The reaction between 2-hydroxy-3,5-difluorophenylboronic acid and 2-substituted 4-bromo-1,3-thiazole involves the substitution of bromine, but the yield of 4-difluoroaryl derivative of 1,3-thiazole is not high (16%) [125].
Acid 3 was also used to produce biaryls and heterobiaryls another method that is implemented as a one-pot conversion of a mixture of 3 with benzene or thiophene in the presence of Mn (III)-acetate, under microwave irradiation (Scheme 48) [126].
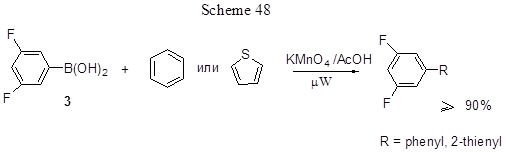
This is the usual radical way to biaryls, and Mn(OAc)3. 2H2O is a one-electron oxidant used to generate radicals, thus leading to the formation of C-C bond. It can be obtained from KMnO4+ CH3COOH under microwave irradiation. The novelty of the proposed method consists in the use of microwave radiation instead of conventional heating, and its advantages are: high product yields, short reaction time, and small consumption of benzene or thiophene, that are to be used for reagents only, but not for solvents.
To prepare the compounds with interesting biomedical properties the synthesis was realized, the initial step of which is Suzuki reaction between a enol triflate of substituted pyrazole and 2,3-difluorophenylboronic acid (5) [113], similar to the reactions of difluorophenylboronic acids 1, 2, 3 and 2,3,5-trifluorophenylboronic acid [113], which runs via the substitution of OTf- group.
In terms of producing materials with interesting liquid-crystal properties the Suzuki reaction was studied between halogenated arenes and acid 5, or its 4-alkoxy and 4-alkyl derivatives, including those with long alkyl chains, resulting in polyfluorinated biphenyls with terminal alkoxy- or alkyl- groups.
The reactions between acid 5 and bromoarenes [84b] or iodoarenes [81, 93] are shown in Scheme 49, while those of its 4-alkoxy derivatives with long alkyl chains are shown in Scheme 50 [81 , 84b, 127, 128].
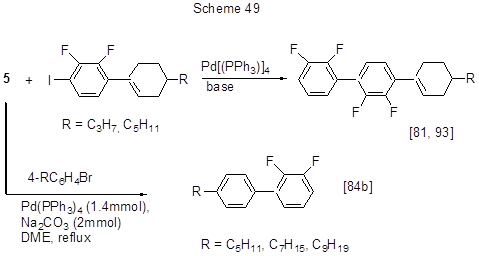
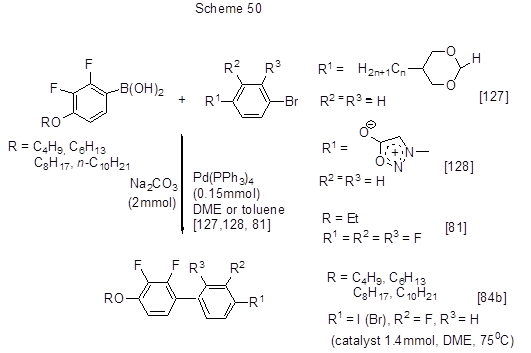
The involvement of 4-alkyl derivatives of acid 5 into the reaction leads to various outcomes. Thus, 2,3-difluoro-4-heptyl-6-methylboronic acid did not react with 2-cyano-4-oxyoctyl-6-methylbromobenzene in the presence of Pd(PPh3)4, and only using of 3 mol% of PdCl2, 6 mol% PPh3, and CsF for base, after careful drying of all reactants, made it possible to produce the corresponding biphenyl with yield 21% (Scheme 51) [85]. In the model experiment with bromobenzene, the yield was 40%.

Cross-coupling of 4-alkyl derivatives of acid 5 either with bromophenol/its ethers [68], or with 1-iodo-2-fluoro-4-bromobenzene is presented in Scheme 52.
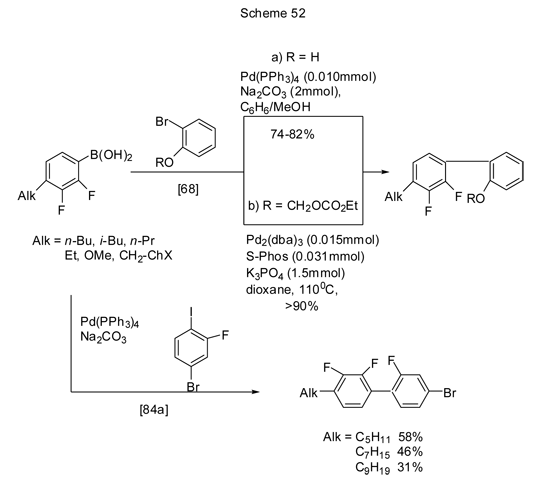
In terms of terphenyl structure design with desired location of substituents, starting from 4-alkyl or 4-aryl acid 5 derivatives, two series of terphenyls were synthesized with terminal alkyl chains containing two fluorines either at their terminal or central ring, wherein in the first case a bromoderivative of biaryl was introduced into the reaction as a halogenated aryl component (Scheme 53) [84a].

Finally, Scheme 54 represents an example of Suzuki reaction with the use ofpara-trimethylsilyl derivative of acid 5 ester resulting in a gradual growth of the chain attached to polyfluorinated quaterphenyls [129]. Since the reactions described elsewhere [129], applied some combinatorial and parallel methods and made use of halogenated derivatives with variable number of fluorines or without fluorines, they resulted in a wide range of biphenyls, ter-, or quaterphenyls containing different number of fluorine atoms. The use of more stable esters of boric acid instead of free acids can reduce the side-process of deborylation. It should be noted that, in examined Suzuki reactions if more than two fluorine atoms occupy the ortho-position to the formed C-C bond, the product yield is low or the reaction does not occur at all.
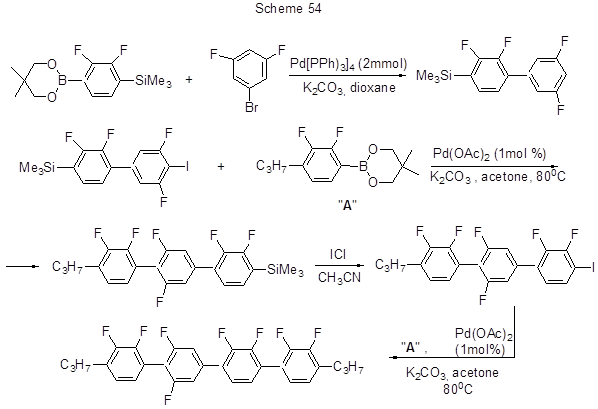
For the formation of biphenyl derivatives in the Suzuki reaction between boric acid 2 and 2-nitro-4-chloromethylbenzoate and o-bromophenol see [130-132], while for that with 3,5-substituted bromobenzene see [133]. The corresponding biphenyl is produced with 94% yield starting with acid 2 and 4-(4'-alkylcyclohexyl)bromobenzene, and using a complex of bis(imino)pyridine Pd (II) as catalyst [134]. Scheme 55 exemplifies the Suzuki reaction between acid 2 and heterocyclic compounds containing bromine and triflate group, and also that between an acid 2 ortho-N-2 pivaloylamide derivative and a triflate belonging to cyclopentene series.

Acid 2 nucleophilic addition to benzaldehyde in the presence of nickel catalyst leads to an oxyalkyl derivative (scheme 56) [139]. The mechanism of transformation is yet unknown, but it is assumed that nickel catalyst acts as Lewis acid.
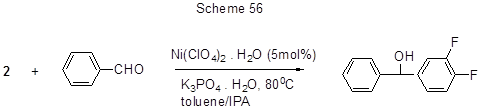
A reaction of heterocyclic trifluoromethanesulfonate similar to that shown in Scheme 55 is realized for phenylboronic acid 4[137]. The same acid enters Suzuki reaction with a chlorinated derivative of substituted 1,3-oxazole [117], p-bromotoluene under microwave irradiation [140], and o-bromoaniline [ 141].
2,6-Difluorophenylboronic acid (6) [104] (just like other isomeric difluorophenylboronic acids), 4-substituted acid 6 [142, 143], and esters of 4-substituted acid 6[123 ] also undergo Suzuki reaction.
2.3.2 Trifluorophenylboronic acids
3,4,5-Trfluorophenylboronic acid (7) undergo Suzuki reaction with 7-substituted 3-iodochromone in the presence of Pd/C and K2CO3, resulting in 80% yield of the corresponding chromone with trifluorophenyl group at position 3 [114, 115]. Reaction involving chlorine atom occurs in the case 7-chloro-4-oxa-2-propyl-1,2,3,4,4a,9,10,10a-octahydrophenantrene, PdCl2(PCy3)2is used as catalyst [144]. Brominated derivatives also readily undergo Suzuki cross-coupling reaction with acid 7. Thus, in the presence of Pd(PPh3)4, (Bu)4NBr and NaHCO3,4-(1-hydroxycyclopentyl-1)bromobenzene and acid 7 react to produce trifluorobiphenyl derivative with 90% yield [145]. 4-(4'-Alkylcyclohexyl) bromobenzene and acid 7 undergo Suzuki reaction with bis(imino)pyridinepalladium (II) complex used as catalyst, the reaction results in the formation of a biphenyl derivative with 4-(4'-alkylcyclohexyl) group on one of its aromatic rings, and three fluorine atoms on its other ring, the reaction yield is 94% [134]. In the case of substituted biphenyl with bromines on each of its aromatic rings [146], or a heterocyclic dibrominated derivative as shown in Scheme 57 [147], the reaction involves both bromine atoms.
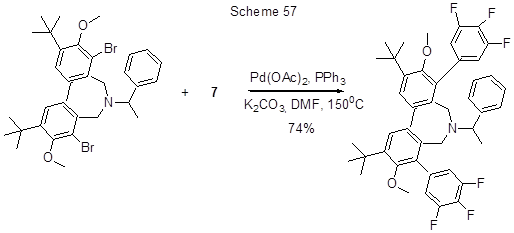
Finally, Suzuki reaction of acid 7 and 6-oxo-8-propyl-7,8,9,10-tetrahydro-6H-benzo[c]-chromen-3-yl trifluoromethanesulfonate with PdCl2(PPh3)2 and aqueous hydrazine hydrate occurs with the participation of OTf-group [148].
2,3,4-Trifluorophenylboronic acid is cross-coupled with o-hydroxybromobenzene [132] to produce compounds with ortho-hydroxybiphenyl systems, those being promising nonpeptide protease inhibitors (accumulation of fluorine atoms contributes to the growth of inhibitor activity). 5-Alkyl- and 5-alkoxy- derivatives of 2,3,4-trifluorophenylboronic acid were added into Suzuki process having in mind to synthesize polyfluorinated terphenyls containing 1,5-disubstituted 2,3,4-trifluorophenyl fragments that have the properties of liquid crystals (Scheme 58) [149].
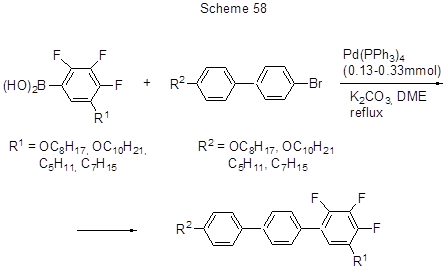
The yields of those reactions are high.
On Suzuki cross-coupling of 2,3,5-trifluorophenylboronic acid with 3-oxocyclohex-1-enyl triflate involving triflate group, see [150].
2.3.3 Pentafluorophenylboronic acid and polyfluorophenylborates
Pentafluorophenylboronic acid is inactive at usual Suzuki cross-coupling conditions, but the use of Ag2O+CsF additives to palladium catalyst promotes the reaction [151]. The reaction is most efficient (yields exceed 90%) when it is carried out with aromatic iodides or bromides, respectively, with C6H5I and XC6H4Br, here X= o-,m-,p-CH3,m-, p-OCH3, p-F, p-OC2H5, p-CF3,p-NO2, 2-naphthyl. In the case of chlorobenzene the yield of pentafluorobiphenyl is only 39%. The best catalytic system for iodides is Pd(PPh3)4/CsF/Ag2O, while that for bromides (and for chlorobenzene) is Pd2(dba)3/P(t-Bu)3/CsF/Ag2O.
Frohn and Bardin [152] exemplify the use of lithium and potassium salts of polyfluorophenylborates as reagents for the manufacture of biphenyl derivatives in the palladium-catalyzed cross-coupling reactions with aromatic iododerivatives in the presence of Ag2O in stoichiometric amounts. The general scheme for those transformations is shown below (Scheme 59).
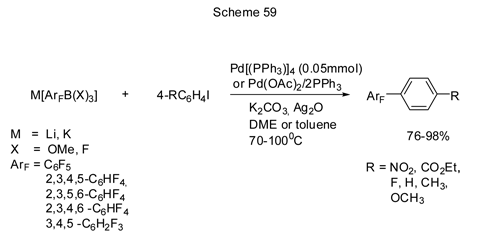
In principle, lithium salts are more reactive than potassium salts, but Ag2O additive equals the reactivity of both salts with respect to this process. Substituted bromobenzenes proved to be less effective than iododerivatives [152 b]. The yield of biphenyl derivatives is also low in the interaction of K[C6F5BF3] with substituted benzodiazoniumtetrafluoroborates [4R-C6H4N2][BF4]. Here Pd(PPh3)4was found to be the best palladium-based catalyst [152c].
The interaction of potassium salt trifluoro-(2,6-difluorophenyl)borate with 2-substituted 5-iodopyridine in the presence PdCl2dppf results in a product of iodine substitution for 2,6-difluorophenyl group, the yield is 17 % [ 153].
2.4. Formation and reactions of the polyfluorinated organozinc and organotin compounds
3-(3,4,5-Trifluorophenyl)-thiophene was synthesized by the Negishi cross-coupling reaction between a zinc complex (produced from 1,2,3-trifluorobenzene) and 3-bromothiophene. The applied catalyst was PdCl2(PPh3)2or Pd(PPh3)4[154]. In [155] to obtain either aryl or heteroaryl zinc reagents from activated aromatics or heteroaromatics with subsequent use of those reagents in Negishi coupling reactions the authors proposed a new, mild and effective base that was 2,2,6,6-tetramethylpiperidinezinc chloride with lithium chloride (TMPZnCl. LiCl), which in its turn was prepared from 2,2,6,6-tetramethylpiperidine, BuLi and ZnCl2 (Scheme 60).
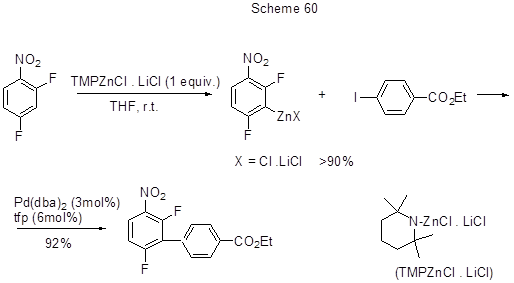
Consequential reactions of 1,3-difluorobenzene or 1,3-difluoro-5-methoxybenzene with p-BuLi and ZnCl2 followed by that with (3-amino-2-bromopyridine-4-yl)-(2,4-difluorophenyl)-methanone in the presence of Pd(PPh3)4 makes it possible to produce difluoroarenes with pyridine fragments (Scheme 61) [112]. It should be noted that the final product with R = H may also be produced by Suzuki reaction using 2,6-difluorophenylboronic acid 6 [112].
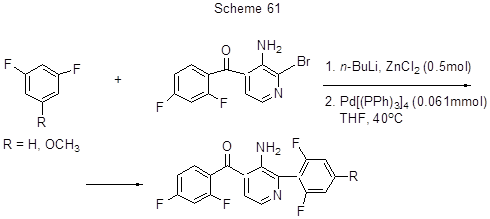
It is assumed that the reaction proceeds via intermediate difluoroarene zinc derivatives that by Negishi reaction with pyridine bromoderivatives are converted to the observed products. Similarly 3,4,5-trifluorophenylation of 4'-(3-n-butylbicyclo[1,1,1]-penta-1-yl)-3,5-difluorobiphenyl at position 4 occurs by reacting it with 3,4,5-trifluorobromobenzene, n-BuLi, ZnCl2and Pd(PPh3)4 [ 156].
The modification of polyfluorinated biphenyls synthesis by Negishi reaction implies preliminary production of polyfluoroaluminumaryl derivatives through direct alumination with an aluminium reagent, and their further trans- metalation with ZnCl2to result in zinc derivatives. Those derivatives enter palladium-catalyzed Negishi cross-coupling with aryl iodides to result in biphenyl derivatives (Scheme 62) [ 157].

One step in the production of biarylcarbamoylindolines, those being promising antidepressants, is the Stille reaction that is the interaction between (2,6- or 3,5-difluorophenyl)tributyltin and 5-bromonicotinate (Scheme 63) [ 158].
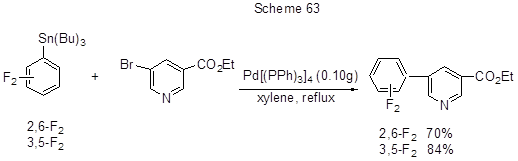
continued in the next issue
List of Abbreviations
1. acac — acetylacetonate
2. AIBN —a,a’ -azobisisobutyrodinitrile
3. BINAP — 2,2’ -bis(diphenylphosphino)-1,1’ - binaphthyl
4. (R)-BINAP — (R)-1,1’ - bis(diphenylphosphino)-2,2’ - binaphthyl
5. Boc — tert-butoxycarbonyl
6. COD — 1,5-cyclooctadiene
7. Cp — cyclopentadienyl
8. Cy — cyclohexyl
9. Cy2NH — N,N-dicyclohexylamine
10. Cyp — cyclopentyl
11. DABCO — 1,4-diazabicyclo[2,2,2]octane
12. davephos — (2-bicyclohexylphosphino)-2’ -(N,N-dimethylamino)biphenyle
13. dba — dibenzylideneacetone
14. Dt BPF — 1,1-bis(di-tert-butylphosphino)ferrocene
15. DiРPF — 1,1’ -bis(di-isopropylphosphino)ferrocene
16. DBU — 1,8-diazabicyclo[5.4.0]undec-7-ene
17. dppe — 1,2-bis(diphenylphosphino)ethane
18. dppf — 1,1’ -bis(diphenylphosphino)ferrocene
19. dppp — 1,3-bis(diphenylphosphino)propane
20. dppbz — 1,2-bis(diphenylphosphino)benzene
21. dii ppbz — 1,2-bis[(4-isopropylphenyl)phosphino]benzene
22. EPHP — 1-ethylpiperidine hypophosphite
23. L — ligand
24. LDA — lithium diisopropylamide
25. LiTMP — lithium tetramethylpiperidide
26. NHC — N-heterocyclic carbene
27. NEM — N-ethylmorpholine
28. OTf — trifluoromethanesulfonate
29. PCy3 - HBF4 — tricyclohexylphosphine tetrafluoroborate
30. Phen — 1,10-phenantroline
31. i-Pr2Im — 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene
32. i-Pr2ImC — 1,3-bis(2,6-diisopropylphenyl)imidazolinium chloride
33. Rh(acac) bis(C2H2) — Rhodium acetylacetonate/bis ethylene
34 PCy2-dmb — 2-(dicyclohexylphosphino)-2’ ,6’ -dimethoxybiphenyl
35. TBAB — tetrabutylammonium bromide
36. TBAF — tetrabutylammonium fluoride
37. TFA — trifluoroacetic acid
38. TMAF — tetramethylammonium fluoride
39. TMEDA — N,N,N’ ,N’ -tetramethylethylenediamine
40. Tf — trifluoromethanesulfonyl
41. tfp — tri(o-furyl)phosphine
42. TMP — 2,2,6,6-tetramethylpyperidine
43. TFSA — trifluoromethanesulphonic acid
44. Ts — tosyl (4-toluenesulphonyl)
References
[2] Morrison R., Boid R. Organicheskaya khimiya., Mir, Moskva, (1974).
[3] Barton D., Ollis V.D. Obshchaya organicheskaya himiya, T.1., Khimiya, Moskva, (1981).
[4] Hassan J., Sevignon M., Gozzi C., Schulz E., Lemair M., Chem. Revs., (2002), 102, 1359-1470.
[5] Ed. Ackermann L. Modern Arylation Methods., John Wiley, Weinheim, (2009).
[6] Smit V.A., Dil'man A.D. Osnovy sovremennogo organicheskogo sinteza., BINOM, Laboratoriya znanij, Moskva, (2009).
[7] Eds. Meijere A., Diederich F. Metal-Catalyzed Cross-Coupling Reactions., John Wiley, Weinheim, (2004).
[8] Schlosser M. Organometallics in Synthesis, 2nd Ed., John Wiley, Chichester, (2002).
[9] Brooke G.M., J. Fluor. Chem., (1997), 86, 1-76.
[10] Filler R., Fiebig Jr. A.E., Mandal B.K., J. Fluor. Chem., (2000), 102, 185-188.
[11] Koda T., Yasntake M., Shinmyozu T., Org. Lett., (2001), 3, 1419.
[12] Tabart M., Picaut G., Desconclois J-F., Dutka-Maben S., Huet Y., Berthaud N., Bioorg. Med. Chem. Lett., (2001), 11, 919-922.
[13] Lundbeck H., Pat. WO 106534 (2009) (C.A., 2009, 151, 337044).
[14] Nakamura S., Sugimoto H., Ohwada T., J. Am. Chem. Soc., (2007), 129, 1724- 1732.
[15] Pat. US 211741 (2006) (C.A., 2006, 145, 377029).
[16] Pat. US 270682 (2006) (C.A., 2007, 146, 27833).
[17]Карпов В.М.,Меженкова Т.В.,Платонов В.Е.,Синяков В.Р.,Щёголева Л.Н.,Ж.орган.хим., (2002), 38, 1210-1217.
[18] Karpov V.M., Meshenkova T.V., Platonov V.E., Sinyakov V.R., J. Fluor. Chem., (2002), 117, 73-81. Karpov V.M., Meshenkova T.V., Platonov V.E., Sinyakov V.R.,Shchegoleva L.N., Russ. J. Org. Chem.,(2002), 38, 1158-1165 (English translation).
[19] Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Y.V., Russ. J. Org. Chem.,(2003),39, 837-842 (English translation).
[20] Zonov Ya.V., Karpov V.M., Platonov V.E., J. Fluor.Chem., (2007), 128, 1058- 1064.
[21] Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Russ. J. Org. Chem.,(2007),43, 1677-1685 (English translation).
[22] Karpov V.M., Mezhenkova T.V., Platonov V.E., Sinyakov V.R., J. Fluor. Chem., (2001), 107, 53-57.
[23] Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Y.V., Russ. J. Org. Chem.,(2006),42, 77-86 (English translation).
[24] a) Bonnefous C., Payne J.E., Roppe J., Zhuang H., Chen X., Severance D., Noble S.A., Smith N.D., Symons K.T., Nguyen P.M., Shiau A.K., Hassig C.A., Sablad M., Rosenkrants N., Zhang Y., Tadimeti S., Wang L., Rix P., Walsh J.P., Yazdani N., J. Med. Chem., (2009), 52, 3047-3062; b) Pat. WO 29592 (2009) (C.A., 2009, 150, 283051); c) Pat. WO 29617 (2009) (C.A., 2009, 150, 282846); d) Pat. WO 29625 (2009) (C.A., 2009, 150, 283049).
[25] Safina L.Yu., Selivanova G.A., Shteingarts V.D., Koltunov K.Yu., Tetrahedron Letters., (2009), 50, 5245-5247.
[26] Inglis S.R., Stojkoski C., Branson K.M., Cawthray J.F., Fritz D., Wiadrowski E., Pyke S.M., Booker G.W., J. Med. Chem., (2004), 47, 5405-5417.
[27] Breault G., Eyemann C.J., Geng B., Momingstar M., Reck F., Pat. WO 134378 (2006) [C.A. (2007), 146, 81779].
[28] Kato T., Saeki K., Kawazoe Y., Hakura A., Mutation Res., (1999), 439, 149- 158. [29] Laev S.S., Gurskaya L.Yu., Selivanova G.A., Beregovaya I.V., Shchegoleva L.N., Vasil’eva N.V., Shakirov M.M., Shteingarts V.D., Eur. J. Org. Chem., (2007), 306-316.
[30] Shuttleworth S.J., Lizarzabaru M.E., Chai A., Coward P., Bioorg. Med. Chem. Letters., (2004), 14, 3037-3042.
[31] Mustafin A.G., Gimadieva A.R., Khalilov I.N., Spirikhin L.N., Fatykhov A.A., Nurushev R.A., Abdrakhmanov I.B., Tolstikov G.A., Bull. Russ. Acad. Sci. Div. Chem.Sci., (1998), 47, 188-190 (English translation).
[32] Gataullin R.R., F. F. Minnigulov F.F., Kudashev A.R., Nurushev R.A., I. B. Abdrakhmanov I.B., Russ. J. Applied Chem., (2002), 75, 95-97 (English translation).
[33] Gataullin R.R., Kazhanova T.V., DavydovaV.A., IsmagilovaA.F., Zarudii F.S., Fatykhov A.A., SpirikhinL.V., I.B. AbdrakhmanovI.B., Russ. J. Chem. Pharm., (1999), 33, , 255-258.
[34] Song Yu., Zhu A., Lv J., Gong G., Xie J., Zhou J., Ye Y., Zhong X., Spectrochim. Acta Part A: Molecular and Biomol. Spectroscopy, (2009), 73, 96-100.
[35] Chackalamannil S., Doller D., Clasty M., Xia Y., Fagen K., Lin Y., Hsung-An Tsai, McPhail A.T., Tetrahedron Letters, (2000), 41, 4043-4047.
[36] Yoshimura H., Kikuchi K., Hibi Sh., Tagami K., Satoh T., Yamauchi T., Ishibahi A., Tai K., Hida T., Tokuhara N., Nagai M., J. Med. Chem., (2000), 43, 2929-2937.
[37] Pat. US 6110959 (2000) (C.A., 1997, 127, 278135); Pat. US 6355669 (2002) (C.A., 1999, 130, 306588); Pat. US 6258811 (2001) (C.A., 1998, 128, 270532).
[38] Pat. US 7045545 (2006) (C.A., 2000, 133, 135218).
[39] Kim B.H., Jeon I., Lee D.B., Park H.J., Jun Y.M., Baik W., J. Fluor. Chem., (2001), 109, 145-149. 106
[40] Yekta S., Krasnova L.B., Mariampillai B., Picard C.J., Chen G., Pandiaraju S., Yudin A.K., J. Fluor. Chem., (2004), 125, 517-525; Pat. WO 51899 (2005) (C.A., 2005, 143, 27355).
[41] Pat. WO 60160 (2009) (C.A., 2009, 150, 313452).
[42] Pat. US 78249 (2003) (C.A., 2001, 134, 86149).
[43] Lafrance M., Rowley Ch.N., Woo T.K., Fagnou K., J. Am. Chem. Soc., (2006), 128, 8754-8756.
[44] Lafrance M., Shore D., Fagnou K., Org. Letters, (2006), 8, 5097-5100.
[45] Youbert N., Urban M., Pohl R., Hocek M., Synthesis, (2008), 1918-1932.
[46] Litvinas N.D., Brodsky B.H., DuBois J., Angew. Chem., Int. Ed.Engl., (2009), 48, 4513-4516.
[47] Zhao D., Wang W., Lian S., Yang F., Lan J., You J., Chemistry – A Europ. J., (2009), 15, 1337-1340.
[48] Itahara T., J. Org. Chem., (1985), 50, 5546-5550.
[49] Ahn J.H., Cho S.Y., Ha J-D., Chu S.Y., Jung S.H., Jung Y.S., Baek J.J., Choi I.K., Shin E.Y., Kang S.L., Kim S.S., Cheon H.G., Yang S.D., Choi J.K., Bioorg. Med. Chem. Letters, (2002), 12, 1941-1946.
[50] Rauf W., Thompson A.L., Brown J.M., Chem. Commun., (2009), 3874-3876.
[51] Laha J.K., Petron Ph., Cuny G.D., J. Org. Chem., (2009), 74, 3152-3155.
[52] Chernyak N., Gevorgyan V., J. Am. Chem. Soc., (2008), 130, 5636-5637; Advanced Synth. and Catalysis, (2009), 351, 1101-1114.
[53] Wei Y., Kan J., Wang M., Li W., Hong M., Org. Letters, (2009), 11, 3346- 3349.
[54] Demchuk O.M., Pietrusiewicz K.M., Synlett, (2009), 18, 1149-1153.
[55] Pat. US 42896 (2009) (C.A., 2009, 150, 237419).
[56] Saito A., Oda S., Fukaya H., Hanzawa Yu., J.Org. Chem., (2009), 74, 1517- 1524.
[57] Nakao Y., Kashihara N., Kanyiva K.S., Hiyama T., J. Am. Chem. Soc., (2008), 130, 16170-16171.
[58] Pat. US 6211375 (2001) (C.A., 1997, 127, 331407).
[59] Pat. US 6136823 (2000) (C.A., 1998, 129, 41087).
[60] Pat. US 6235764 (2001) (C.A., 2000, 132, 22961).
[61] Pat. US 6348624 (2002) (C.A., 1999, 131, 73434); Pat. US 6329391 (2001) (C.A., 2002, 136, 20031).
[62] Pat. EP 1223169 (2002) (C.A., 1996, 125, 142540).
[63] Pat. US 114666 (2003) (C.A., 2003, 138, 55976).
[64] Chodakowski J., Klis’ T., Servatowski J., Tetrahedron Letters, (2005), 46, 1963-1965.
[65] Anquetin G., Greiner J., Vierling P., Tetrahedron, (2005), 61, 8394-8404.
[66] Omura K., Uchida T., Irie R., Katsuki T., Chem. Commun., (2004), 2060- 2061.
[67] Leroux F., Hutsehenreuter T.U., Charriere C., Scopelliti R., Hartman R.W., Helv. Chim. Acta, (2003), 86, 2671-2686.
[68] Inagaki H., Tsuruoka H., Hornsby M., Lesley S.A., Spraggon G., Ellman J.A., J. Med. Chem., (2007), 50, 2693-2699.
[69] Bashore C.G., Vetelino M.G., Wirtz M.C., Brooks P.R., Frost H.N., McDermort R.E., Whritenour D.C., Ragan J.A., Rutherford J.L., Makowski T.W., Brenek S. J., Coe J.W., Org. Letters, (2006), 8, 5947-5950.
[70] Becht J.M., Ngouela S., Wagner A., Mioskowski Ch., Terahedron, (2004), 60, 6853-6858.
[71] Leroux F.R., Bonnafoux L., Heiss C., Colobert F., Lanfranchi D.A., Advanced Synth. and Catalysis, (2007), 349, 2705-2713.
[72] Milne J.E., Buchwald S.L., J. Am. Chem. Soc., (2004), 126, 13028-13032.
[73] Heis C., Leroux F., Schlosser M., Eur. J. Org. Chem., (2005), 5242-5247.
[74] Pat. US 6388146 (2002) (C.A., 1999, 131, 163453).
[75] Scott J.P., Brower S.E., Davis A.J., Brands K.M.J., Synlett, (2004), 1646-1648.
[76] Davies A.J., Scott J.P., Bishop B.C., Brands K.M., Brewer S.E., DaSilva J.O., Dormer P.G., Dolling U-H., Gibb A.D., Hammond D.C., Lieberman D.D., Palucki M., Payack J.T., J. Org. Chem., (2007), 72, 4864-4871.
[77] Pat. US 42851 (2009) (C.A., 2007, 147, 234875).
[78] Nishida J., Fujiwara Yu., Yamashita Y., Org. Letters, (2009), 11, 1813-1816.
[79] Pat. US 131482 (2009) (C.A., 2009, 150, 563826).
[80] Chen Sh., Huby N.J.S., Kong N., Moliterni J.A., Jonh A., Morales O.J., Pat. US 170920 (2009) (C.A., 2009, 151, 123990).
[81] Keum H.-W., Roh S.D., Do Y.-S., Lee J.-H., Kim Y.-B., Mol. Crystals and Liquid Crystals, (2005), 439, 189-199.
[82] Bremer M., Lietrau L., New J. Chem., (2005), 29, 72-74.
[83] Pat. US 247585 (2007) (C.A., 2007, 147, 494408).
[84] a) Glendenning M.E., Goodby J.W., Hird M., Toyne K.J., J. Chem. Soc., P2, (2000), 27-34; b) J. Chem. Soc., P2 (1999), 481-491.
[85] Cammidge A.N., Crepy K.V.L., J. Org. Chem., (2003), 68, 6832-6835. [86] Popov I., Do H-Q., Daugulis O., J. Org. Chem., (2009), 74, 8309-8313.
[87] Coe P.L., Rees A.J., J. Fluor. Chem., (2000), 101, 45-60.
[88] Do H.-Qu., Daugulis O., J. Am. Chem. Soc., (2008), 130, 1128-1129.
[89] Do H.-Qu., Khaw R.M.K., Daugulis O., J. Am. Chem. Soc., (2008), 130, 15185-15192.
[90] Pat. US 76266 (2009) (C.A., 2009, 150, 329777).
[91] Do H.-Qu., Daugulis O., Chem. Commun., (2009), 6433-6435.
[92] Matsuyama N., Kitahara M., Hirano K., Satoh T., Miura M., Org. Letters, (2010), 12, 2358-2361.
[93] Susuki A., J. Organomet. Chem., (1999), 576, 147-168.
[94] Molander G.A., Biolatto B., J. Org. Chem., (2003), 68, 4302-4314.
[95] Kotsikorou E., Song Y., Chan J.M.W., Faelens S., Tovian Z., Broderick E., Bakalara N., Docampo R., Oldfield E., J. Med. Chem., (2005), 48, 6128-6139.
[96] Song Y., Oldfield E., Lin F.-Y., Yin F, Mikkamala D., Dushyant C., Cao R., Hensler M., Nizet V., Poveda C-A.R., Pacanowska D.G., Wang H., Morita C.T., J. Med. Chem., (2009), 52, 976-988.
[97] Kumar M., Kaur K., Sinha S., Gupta S., Palle V., Chugh A., Pat US 12116 (2009) (C.A., 2007, 146, 162865).
[98] Feuerstein M., Berthiol F., Doucet H., Santelli M., Synlett, (2002), 1807-1810.
[99] Kulmaelae T., Kuuloja N., Franzen R.X., Rissanen R.K., Europ. J. Org. Chem., (2008), 4019-4024.
[100] Guo M., Zhang Q., Tetrahedron Letters, (2009), 50, 1965-1968.
[101] Duplantier A.J., Kraus K.G., Lu J., Noha M., Rogers B.N., Zhang L., Efremov I., Candler J., O’ Sillivan T.J., Ganong A.H., Hanks A.N., Lazzaro Jr. J.T., McCarthy S.A., Siuciak J.A., Doran A.C., Haas J.A., Spracklin D.K., Bioorg. Med. Chem. Letters, (2009), 19, 2524-2529.
[102] Pat. EP 1842854 (2007) (C.A., 2007, 147, 427511); Pat. WO 148829 (2008) (C.A., 2009, 150, 44027); Pat. US 200920 (2009) (C.A., 2007, 146, 430964).
[103] Pat. WO 11447 (2009) (C.A., 2009, 150, 121792).
[104] Pat. US 163508 (2009) (C.A., 2009, 150, 447978). 109
[105] Pat. WO 112854 (2009) (C.A., 2009, 151, 392217).
[106] Pat. WO 17664 (2009) (C.A., 2009, 150, 191528); Pat. WO 10150 (2010) (C.A., 2010, 152, 215298).
[107] Pat. US 203705 (2009) (C.A., 2009, 151, 245649).
[108] Pat. WO 155388 (2009) (C.A., 2010, 152, 97501); Pat. WO 155389 (2009) (C.A., 2010, 152, 97472).
[109] Pat. WO 102460 (2009) (C.A., 2009, 151, 289162).
[110] Pat. WO 153285 (2009) (C.A., 2010, 152, 97447).
[111] Ce G., Guo H., He J., Wang F., Zou D., J. Organomet. Chem., (2009), 694, 3050-3057.
[112] Lumeras W., Caturla F., Vidal L., Esteve C., Vidal B., Balaque C., Orellana A., Godessard N., Dominguez M., Roca R., Huerta J.M., J. Med. Chem., (2009), 52, 5531-5545.
[113] Imbriglio J.E., Chang S., Liang R., Raghavan S., Schmidt D., Smenton A., Tria S., Schrader T.O., Jung J.K., Esser C., Taggart A.K.P., Cheng K., Carballo-Jane E., Waters M.G., Tata J.P., Colletti S.L., Bioorg. Med. Chem. Letters, (2009), 19, 2121-2124.
[114] Matin A., Gavandl N., Kim M.S., Yang N.X., Salam N.K., Hanzahan J-R., Roubin R.H., Hibbs D.E., J. Med. Chem., (2009), 52, 6835-6850.
[115] Pat. WO 26657 (2009) (C.A., 2009, 150, 306367).
[116] Hagdar S.N., Comery T.A., Dunlop J., Ghiron Ch., Bettinetti L., Bochmann H., LaPosa S., Micco I., Pollastrini M., Quinn J., Roncarati R., Scali C., Valacchi M., Varrone M., Zanaletti R., Bioorg. Med. Chem., (2009), 17, 5247-5258.
[117] Lee S., Yi K.Y., Youn S.J., Lee B.A., Yoo S., Bioorg. Med. Chem. Letters, (2009), 19, 1329-1331.
[118] Pat. WO 52341 (2009) (C.A., 2009, 150, 472421).
[119] Blake T.D., Hamper B.C., Huang W., James R., Moon J.B., Neel B.E., Olson K.L., Pelc M.J., Schneitzer B.A., Thorarensen A., Trujillo J.I., Turner S.R., Pat. US 146569 (2008) (C.A., 2008, 149, 79492).
[120] Pat. US 170907 (2009) (C.A., 2007, 146, 62449).
[121] Burns M.J., Fairlamb I.J.S., Kapdi A.R., Sehnal P., Taylor R.J.K., Org. Letters, (2007), 9 , 5397-5400; Fairlamb I.J.S., Sehnal P., Taylor R.J.K., Synthesis, (2009), 508-510.
[122] Pat. EP 2123623 (2009) (C.A., 2008, 149, 235600).
[123] a) Pat WO 120789 (2009) (C.A., 2009, 151, 403339); b) Pat. EP 2116522 (2009) (C.A., 2008, 149, 343403).
[124] Li G., Stamford A.W., Huang Y., Cheng K.Ch., Cook J., Farley C., Gao J., Ghibaudi L., Greenlee W.J., Guzzi M., van Heek M., Hwa J.J., Kelly J., Mullins D., Parker E.M., Wainhaus S., Zhang X., Bioorg. Med. Chem. Letters, (2008), 18, 1146- 1150.
[125] Bouillot A.M., Dodic N., Gellibert F.J., Mirguet O., Pat. WO 15652 (2010) (C.A., 2010, 152, 238948).
[126] Demir A.S., Findik H., Saygili N., Subari N.T., Tetrahedron, (2010), 66, 1308-1312.
[127] Dong Ch.Ch., Styring P., Goodby J.W., Chan L.K.M., J. Mater. Chem., (1999), 9, 1669-1678.
[128] Yelamaggad C.V., Mathews M., Hizemath U.S., Rao D.S., Prasad S.K., Tetrahedron Letters, (2005), 46, 2623-2626.
[129] Deeg O., Bauerle P., Org. and Biomol. Chem., (2003), 1, 1609-1624.
[130] Pat. WO 52722 (2006) (C.A., 2006, 144, 488936).
[131] Tomson S.A., Banker P., Bickett D.M., Boucheron J.A., Carter H.L., Clancy D.C., Cooper J.P., Dickerson S.H., Garrido D.M., Nolte R.T., Peat A.J., Sheckler L.R., Sparks S.M., Tavares F.X., Wang L., Wang T.Y., Weiel J.E., Bioorg. Med. Chem. Letters, (2009), 19, 1177-1182.
[132] Wood W.J.L., Patterson A.W., Tsuruoka H., Jain R.K., Ellman J.A., J. Am. Chem. Soc., (2005), 127, 15521-15527.
[133] Pat. WO 77385 (2009) (C.A., 2009, 151, 77775).
[134] Liu P., Zhou L., Li X., He R., J. Organomet. Chem., (2009), 694, 2290-2294.
[135] Bey E., Marchais-Oberwinkler S., Negri M., Kruchten P., Oster A., Kllin T., Spadaro A., Werth R., Frotscher M., Birk B., Hartmann R.W., J. Med. Chem., (2009), 52, 6724-6743.
[136] Pat. WO 45383 (2009) (C.A., 2009, 150, 396692).
[137] Pat. US 258866 (2009) (C.A., 2008, 148, 17746).
[138] Jaroch S., Hölscher P., Rehwinkel H., Sülzle D., Burton G., Hillmann M., McDonald F.M., Bioorg. Med. Chem. Letters, (2002), 12, 2561-2564; Jaroch S., Rehwinkel H., Hölscher P., Sülzle D., Burton G., Hillmann M., McDonald F.M., Miklautz H, Bioorg. Med. Chem. Letters, (2004), 14, 743-746.
[139] Zhou L., Du X., He R., Ci Z., Bao M., Tetrahedron Letters, (2009), 50, 406- 408.
[140] Stencel L.M., Kormos C.M., Avery K.S., Leedbeater N.E., Org. Biomol. Chem., (2009), 7, 2452-2457.
[141] Pat. US 239748 (2009) (C.A., 2007, 146, 121670).
[142] Pat. EP 2011788 (2009) (C.A., 2007, 147, 502372).
[143] Barberousse V., Bondoux M., Tomas D., Peyrou V., Pat. US 293768 (2008) (C.A., 2006, 145, 377529); Bondoux M., Mignon L., Ou K., Renaut P., Tomas D., Barberousse V., Tetrahedron Letters, (2009), 50, 3872-3876.
[144] Eidenschink R., Kretzchmann H., Pat. US 131689 (2009) (C.A., 2007, 147, 200299).
[145] Pat. WO 91884 (2009) (C.A., 2009, 151, 208832).
[146] Pat. US 29935 (2010) (C.A., 2008, 148, 403569).
[147] Lugo B., Allbutt B., Beaumont D., Butt U., James A.R., Synlett, (2009), 675- 680.
[148] Taugerbeck A., Montenegro E., Manabe A., Plach H., Pat US 247620 (2009) (C.A., 2007, 147, 177293).
[149] Hird M., Goodby J.W., Gough N., Toyne K.J., J. Mater. Chem., (2001), 11, 2732-2742.
[150] Raghavan S., Schmidt D.R., Darby R., Steven S.L., Abi- Smenton A.L., Pat. US 62269 (2009) (C.A., 2007, 147, 277179).
[151] Korenaga T., Kosaki T., Fukumura R., Ema T., Sakai T., Org. Letters, (2005), 7, 4915-4917.
[152] a) Frohn H-J., Adonin N. Yu., Bardin V.V., Starichenko V.F., J. Fluor. Chem., (2003), 122, 195-199; b) Tetrahedron Letters, (2002), 43, 8111-8114; c) J. Fluor. Chem., (2002), 117, 115-120.
[153] Pat. WO 109743 (2009) (C.A., 2009, 151, 337021).
[154] Levi M.D., Gofer Y., Cherkinsky M., Birsa M.L., Aurbach D., Berlin A., Phys. Chem. Chem. Phys., (2003), 5, 2886-2893.
[155] Mosrin M., Knochel P., Org. Letters, (2009), 11, 1837-1840.
[156] Pat. EP 987238 (2000) (C.A., 1998, 129, 223634).
[157] Wunderlich S.H., Knochel P., Angew. Chem., Int. Ed.Engl., (2009), 48, 1501- 1504.
[158] Bromidge S.M., Dabts S., Davies D.T., Davies S., Duckworth D.M., Forbes I.T., Gaster L.M., Ham P., Jones G.E., King F.D., Mulholand K.R., Saunders D.V., Wyman P.A., Blaney F.E., Clarke S.E., Blackburn T.P., Holland V., Kennett G.A., Lightower S., Middlemiss D.N., Trail B., Rilly G.J., Wood M.D., J. Med. Chem., (2000), 43, 1123-1134. [158a] Pat. WO 14268 (2009), (C.A., (2009), 150, 213983).
[159] a) Hatakeyama T., Hashimoto S., Ishizuka K., Nakamura M., J. Am. Chem. Soc., (2009), 131, 11949-11963; b) Hatakeyama T., Nakamura M., J. Am. Chem. Soc., (2007), 129, 9844-9845.
[160] Mayer M., Czaplik W.M., Jacobi von Wangelin A., Synlett, (2009), 2919- 2923.
[161] Ok H.O., Reigle L.B., Candelore M.R., Cascieri M.A., Colwell L.F., Deng L., Feeney W.P., Forrest M.J., Hom G.J., MacIntyre D.E., Stader C.D., Tota L., Wang P., Wyvratt M.J., Fisher M.H., Weber A.E., Bioorg. Med. Chem. Letters, (2000), 10, 1531-1534.
[162] Hatakeyama T., Kondo Y., Fujiwara Yu-J., Takaya H., Nakamura M., Ito Sh., Nakamura E., Chem. Commun., (2009), 1216-1218.
[163] Van Huis Ch.A., Casimiro-Garcia A., Bigge C.F., Cody W.L., Dudley D.A., Filipski K-J., Heemstra R.J., Kohrt J.T., Leadley Jr. R.J., Narasimhan L.S., McClanahan T., Mochalkin I., Pamment M., Thomas P.T., Sahasrabudhe V., Schaum R.P., Edmunds J.J., J. Bioorg. Med. Chem., (2009), 17, 2501-2511.
[164] Pat. WO 13126 (2009) (C.A., 2009, 150, 191522); Orsini P., Menichin-Cheri M., Vanotti E., Panzeri A., Tetrahedron Letters, (2009), 50, 3098-3100.
[165] Pat. WO 70869 (2009) (C.A., 2009, 151, 33423).
[166] Davies A.J., Scott J.P., Bishop B.C., Brands K.M., Brewer S.E., DaSilva J.O., Dormer P.G., Dolling U-H., Gibb A.D., Hammond D.C., Lieberman D.D., Palucki M., Payack J.T., J. Org. Chem., (2007), 72, 4864-4871.
[167] Pat. WO 140128 (2009) (C.A., 2009, 151, 571119).
[168] Abarbri M., Dehmel F., Knochel P., Tetrahedron Letters, (1999), 40, 7449- 7454.
[169]. ZAO NPO «Pim-Invest». Sintezy ftororganicheskih soedinenij., Izd.«Trovant», Moskva, (2005), c. 123, 124.
[170] Scheuermann G.M., Steurer P., Muelhaupt R., Rumi L., Bannwarth W., J. Am. Chem. Soc., (2009), 131, 8262-8270.
[171] Zhang X., Mu F., Robinson B., Wang P., Tetrahedron Letters, (2010), 51, 600-601.
[172] Audia J.E., Mergott D.J., Sheehan S.M., Watson B.M., Pat. US 275566 (2009) (C.A., 2009, 151, 528778).
[173] Pat. WO 103007 (2009) (C.A., 2009, 151, 288987).
[174] Gordeev M.F., Pat US 48305 (2009) (C.A., 2009, 150, 214368).
[175] Pulici M., Zuccotto F., Badari A., Nuvoloni S., Cervi G., Traquandi G., Biondaro S., Trifiro P., Marchionni C., Modugno M., Pat. WO 10154 (2010) (C.A., (2010), 152, 215278).
[176] Liu J., Jian T., Guo L., Atanasova T., Nargund R.P., Tetrahedron Letters, (2009), 50. 5228-5230.
[177] Heiss C., Schlosser M., Eur. J. Org. Chem., (2003), 447-451.
[178] Morgan B.P., Galdamez G.A., Gilliard Jr. R.J., Smith R.C., Dalton Trans., (2009), 2020-2028.
[179] a) Organ M.G., Calimsiz S., Sayah M., Hoi K.H., Lough A.J., Angew. Chem., Int. Ed.Engl., (2009), 48, 2383-2387; b) Altenhoft G., Goddard R., Lehmann Ch.W., Glorius F., J. Am. Chem. Soc., (2004), 126, 15195-15201.
[180] Sakamoto Y., Suzuki T., Miura A., Fujikawa H., Tokito S., Taga Y., J. Am. Chem. Soc., (2000), 122, 1832-1833.
[181] Yudin A.K., Martyn L.J.P., Pandiaraju S., Zheng J., Lough A., Org. Letters, (2000), 2, 41-44.
[182] Pat. WO 111299 (2009) (C.A., 2009, 151, 358919).
[183] Burton D.J., Hartgraves G.A., J. Fluor.Chem., (2009), 130, 254-258.
[184] Vinigradov A.S., Krasnov V.I., Platonov V.E., Russ. J. Org. Chem., (2008), 44, 95-102 (English translation).
[185] Hori A., Kataoka H., Akacaka A., Okano T., Fujita M., J. Polymer Sci., part A: Polymer Chem., (2003), 41, 3478-3485.
[186] Facchetti A., Yoon M-H., Stern Ch.L., Katz H.E., Marks T.J., Angew. Chem., Int. Ed. Engl., (2003), 42, 3900-3903. 114
[187] Cho D.M., Parkin S.R., Watson M.D., Org. Letters, (2005), 7, 1067-1068.
[188] Blass B.E., Huang C.T., Kawamoto R.M., Li M., Liu S., Portlock D.E., Rennells W.M., Simmons M., Bioorg. Med. Chem. Letters, (2000), 10, 1543-1546.
[189] Ragni R., Orselli E., Kottas G.S., Omar O.H., Babudri F., Pedone A., Naso F., Farinola G., DeCola L., Chemistry – A Europ. J., (2009), 15, 136-148.
[190] Nagao I., Shimizu M., Hiyama T., Angew. Chem., Int. Ed. Engl., (2009), 48, 7573-7576.
[191] Frattarelli D., Ratner M.A., Marks T.J., Schiavo M., Facchetti A., J. Am. Chem. Soc., (2009), 131, 12595-12612.
[192] Pat. WO 106577 (2009) (C.A., 2009, 151, 313566).
[193] Pat. EP 1674452 (2006) (C.A., 2005, 142, 355051); Pat. EP 1780197 (2007) (yield 83%) (C.A., 2006, 144, 191974); Tecle H., Shao J., Li Y., Kuthe M., Kazmirski S., Penzotti J., Ding Y-H, Moshinsky D., Coli R., Jhawar N., Bora E., JaquesO’ Hagan S., Wu J., Ohren J., Bioorg. Med. Chem. Letters, (2009), 19, 226-229 (yield 65%).
[194] Wang C.H., Alluri S., Ganguly A.K., Tetrahedron Letters, (2009), 50, 1879- 1881.
[195] Matsuda Sh., Takahashi M., Monguchi D., Mori A., Synlett, (2009), 1941- 1944.
[196] Huang Ch., Gevorgyan V., J. Am. Chem. Soc., (2009), 131, 10844-10845.
[197] Geramita K., McBee J., Tilley T.D., J. Org. Chem., (2009), 74, 820-829.
[198] Marchetti F., Pampaloni G., Passarelli V., Masi F., Sommazzi A., Spera S., J. Fluor. Chem., (2009), 130, 341-347.
[199] Pat. WO 117097 (2009) (C.A., 2009, 151, 403303).
[200] Baumann K., Flohr A., Goetschi E., Jacobsen H., Jolidon S., Luebbers T., Pat. US 181965 (2009) (C.A., 2009, 151, 148313).
[201] Oswald C.L., Peterson J.A., Lam H.W., Org. Letters, (2009), 11, 4504-4507.
[202] Mariaca R., Labat G., Behrnd N-R., Bonin M., Helbling F., Eggli P., Couderc G., Hulliger J., Neels A., Stoeckli-Evans H., J. Fluor. Chem., (2009), 130, 175-196.
[203] Jayanth T.T., Zhang L., Johnson T.S., Malinakova H.C., Org. Letters, (2009), 11, 815-818.
[204] Takayuki T., Yuhei Y., Masashi K., Toshiaki M., Org. Letters. (2009), 11, 1043-1045.
[205] Martin R.E., Wytko J.A., Diederich F., Helv. Chim. Acta, (1999), 82, 1470- 1479.
[206] Rosenblum S.B., Huynh T., Afonso A., Davis Jr. H.R., Tetrahedron, (2000), 56, 5735-5742.
[207] Pat. US 42841 (2009) (C.A., 2009, 150, 168179).
[208] Lledo A., Restorp P., Rebeck Jr. J., J. Am. Chem. Soc., (2009), 131, 2440- 2444.
[209] a) Isaac M., Slassi A., Silva K.D., Xin T., Tetrahedron Letters, (2001), 42, 2957-2960; b) Ying M., Smentek M.G., Day C.S., Welker M.E., Ma R., Torti S.V., Letters in Org. Chem., (2009), 6, 242-251.
[210] Matsnev A., Noritake S., Nomura Y., Tokunaga E., Nakamura S., Shibata N., Angew. Chem., Int. Ed. Engl., (2010), 49, 572-576.
[211]. Biffis A., Scattolin E., Ravasio N., Zaccheria F., Tetrahedron Letters, (2007), 48, 8761-8764; Luque R., MacQuarrie D.J., Org. Biomol. Chem., (2009), 7, 1627- 1632; Johnson S.A., Liu F-Q., Suh M.C., Zuercher S, Haufe M., Mao S.S.H., Tilley T.D., J. Am. Chem. Soc., (2003), 125, 4199-4211; Zeidan T.A., Kovalenko S.V., Monoharan M., Clark R.J., Ghiviriga J., Alabugin I.V., J. Am. Chem. Soc., (2005), 127, 4270-4285; Smith C.E., Smith P.S., Thomas R.L., Robins E.G., Collings J.C., Dai Ch., Scott A.J., Borwick S., Batsanov A.S., Watt S., Clark C.J., VineyCh., Howard J.A.K., Clegg W., Marder T.B., J. Mater. Chem., (2004), 14, 413-420.
[212] a) Ojima I. Fluorine in Medicinal Chemistry and Chemical Biology, J. Wiley Publication , Willy-Blackwell, (2009), p. 299, 301; b) Benmansour H., Chambers R.D., Sandford G., Yufit D.S., Hovard J.A.K., Arkivoc, (2007), (XI), 46-55.
[213] Amii H., Uneyama K., Chem. Revs., (2009), 109, 2119-2183.
[214] Deck P.A., Kroll C.E., Hollis W.G., Fronczek F.R., J. Organomet. Chem., (2001), 637-639, 107-115.
[215] Deck P.A., McCauley B., Slebodnick C., J. Organomet. Chem., (2006), 691, 1973-1983.
[216] Gurjar M., Reddy D.S., Murugaiah A., Murugaiah S., Synthesis, (2000), 1659- 1661.
[217] [212a, p. 307].
[218] Sasaki Sh., Tanabe Y., Yoshifuji M., Bull. Chem. Soc. Japan, (1999), 72, 563- 572.
[219] Morrison D.J., Trefz T.K., Piers W.E., McDonald R., Parvez M., J. Org. Chem., (2005), 70, 5309-5312.
[220] Yang Zh-Yu., Burton D.J., J. Fluor. Chem., (2000), 102, 89-103.
[221] Wang Y., Parkin S.R., Giersehner J., Watson M.D., Org. Letters, (2008), 10, 3307-3310.
[222] Wood T.K., Waren W.E., Keay B.A., Parvez M., Angew. Chem., Int. Ed. Engl., (2009), 48, 4009-4012.
[223] Nitschke J. K., Tilley T.D., J. Am. Chem. Soc., (2001), 123, 10183-10190.
[224] Zahn A., Brotschi C., Leumann C.J., Chemistry – A Europ. J., (2005), 11, 2125-2129.
[225] Pat. WO 93467 (2006) (C.A., 2006, 145, 326323).
[226] Ishihara K., Hasegawa A., Yamamoto H., Angew. Chem., Int. Ed. Engl., (2001), 40, 4077-4079.
[227] Crouch D.J., Skabata P.J., Heeney M., Culloch I., Coles S.J., Hursthouse M.B., Chem. Commun., (2005), 1465-1467.
[228] Wang Y., Parkin S.R., Watson M.D., Org. Letters, (2008), 10, 4421-4424.
[229] Deck P.A., Lane M.J., Montgomery J.K., Slebodnick C., Frouczek F.R., Organometallics, (2000), 19, 1013-1024.
[230] Caster K.C., Keck Ch. G., Walls R.D., J. Org. Chem., (2001), 66, 2932-2936.
[231] Schlosser M., Guio L., Leroux F., J. Am. Chem. Soc., (2001), 123, 3822- 3823.
[232] Coe J.W., Wirtz M.C., Bashore C.G., Candler J., Org. Letters, (2004), 6, 1589- 1592.
[233] Pat. WO 128142 (2006) (C.A., 2007, 146, 27723).
[234] Pat. US 6362365 (2002) (C.A., 2000, 132, 308136).
[235] Tang G., Yang Y.G., Wen J.X., Mol. Cryst. Liquid Cryst. Science and Technology. Sect. A: Mol. Cryst. and Liquid Cryst., (2002), 373, 17-24.
[236] Pat WO 40131 (2006) (C.A., 2006, 144, 390561).
[237] Nuritomi M., Murofushi H., Nakashima N., Bull. Chem. Soc. Japan, (2004), 77, 2121-2128.
[238] Dutta T., Woody K.B., Watson M.D., J. Am. Chem. Soc., (2008), 130, 452- 453.
[239] Yan Sh-J., Huang Ch., Zeng X-H., Huang R., Lin J., Bioorg Med. Chem. Letters, (2010), 20, 48-51. 117
[240] Mallach E., Kuhn N., Maichle-Moessmer C., Steimann M., Stroebele M., Zeller K-P., Z. Naturforsch. B. Chemical Sciences, (2009), 64, 1176-1182.
[241] Pat. US 5856557 (1999) (C.A., 1998, 128, 88679).
[242]. Bella M., Kobbelgaard S., Jørgensen K.A., J. Am. Chem. Soc., (2005), 127, 3670-3671.
[243] Kobbelgaard S., Bella M., Jørgensen K.A., J. Org. Chem., (2006), 71, 4980- 4987.
[244] Steffen A., Sladek M.J., Braun T., Neumann B., Stammler H.G., Organometallics, (2005), 24, 4057-4064.
[245] Schaub T., Backes M., Radius U., J. Am. Chem. Soc., (2006), 128, 15964- 15965.
[246] Braun T., Perutz R.N., Sladek M.I., Chem. Commun., (2001), 2254-2255.
[247] Yoshikai N., Mashima H., Nakamura E., J. Am. Chem. Soc., (2005), 127, 17978-17979.
[248] Wang J-R., Manabe K., Org. Letters, (2009), 11, 741-744.
[249] Ishikawa S., Manabe K., Synthesis, (2008), 3180-3182; Manabe K., Ishikawa S., Synthesis, (2008), 2645-2649.
[250] Braun T., Isundu J, Steffen A., Neumann B., Stammler H-G., Dalton Trans., (2006), 5118-5123.
[251] Guo H., Kong F., Kanno K., He J., Nakajima K., Takahashi T., Organometallics, (2006), 25, 2045-2048.
[252] Korn T.J., Schade M.A., Cheemala M.N., Wirth S., Guevata S.A., Cahiez G., Synthesis, (2006), 3547-3574; Korn T.J., Schade M.A., Wirth S., Knochel P., Org. Letters, (2006), 8, 725-728.
[253] Edelbach B.L., Kraft B.M., Jones W.D., J. Am. Chem. Soc., (1999), 121, 10327-10331.
[254] Miller A.O., Krasnov V.I., Peters D., Platonov V.E., Miethchen R., Tetrahedron Letters, (2000), 41, 3817-3819.
[255] Buckley H.L., Sun A.D., Love J.A., Organometallics, (2009), 28, 6622-6624; Wang T., Love J.A., Organometallics, (2008), 27, 3290-3296.
[256] Blay G., Fernandez I., Monleon A., Pedro J-R., Vila C., Org. Letters, (2009), 11, 441-444. 118
[257] Musso D.L., Cochran F.R., Kelley J.L., McLean Ed.W., Selph J.L., Rigdon G.C., Orr G. Faye, Davis R.G., Cooper B.R., Styles V.L., Thompson J.B., Hall W.R., J. Med. Chem., (2003), 46, 399-408.
[258] Tagat J.R., McCombic S.W., Nazareno D.V., Boyll C.D., Kozlowski J.A., Chackalamannil S., Josien H., Wang Ya., Zhou G., J. Org. Chem., (2002), 67, 1171- 1177.
[259] Andrus M.B., Liu J., Tetrahedron Letters, (2006), 47, 5811-5814; Moran B.N., Anderson F.P., Cloonan S., Butler N.E., Kenny P.T.M., Deverg A., Verughese S., Draper S.M., Bioorg. Med. Chem., (2009), 17, 4510-4522.
[260] Cornella J., Lu P., Larrosa I., Org. Letters, (2009), 11, 5506-5509.
[261] Shang R., Fu Y., Wang Y., Xu Q., Yu H-Z., Liu L., Angew. Chem., Int. Ed. Engl., (2009), 48, 9350-9354.
[262] Wyatt P., Hudsen A., Charmant J., Orpen A.G., Phetmung H., Org. Biomol. Chem., (2006), 4, 2218-2232.
[263] Iovel I, Mertins K., Kirschel J., Zapf A., Beller M., Angew. Chem., Int. Ed. Engl., (2005), 44, 3913-3917.
[264] Li L., Chen H., Lin Y., Synth. Commun., (2007), 37, 985-991.
Recommended for publication by prof. V.E. Platonov
Fluorine Notes, 2014, 92, 1-2
