Received: December , 2013
Fluorine Notes, 2014, 92, 3-4
SYNTHESIS OF NOVEL DERIVATIVES OF "FLOROKSAN" PLANT GROWTH STIMULANT
O.Yu. Federovsky, I.S. Schelik, N.D. Kagramanov, N.D. Chkanikov
A.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova
28, V-334, GSP-1, 119991 Moscow, Russia
e-mail: offskii@rambler.ru
Abstract: Reported is the preparation of some novel derivatives of "Floroksan" that is a fluorinated plant growth regulator.
Keywords: organofluoric substances, plant growth regulator, "floroksan", "floroksan" derivatives, transformations of substituted 3,3,3-trifluoropropionic acid.
The search for novel plant growth regulators is an actual challenge to chemist engaged in organic synthesis [1]. "Floroksan" (1) is a fluorinated substance currently being introduced in practical agriculture as a promising plant growth stimulator [2]. The benefits that will accrue to it are extremely small active concentration, low toxicity, easy biodegradation, and hence negligible environment impact combined with efficient plant growth stimulation [3].
Here we are reporting the synthesis of some derivatives of (1), conducted in order to produce even more efficient products. The three types of thus conducted chemical transformations involved phenyl rings, ester groups, and hydroxyl groups correspondingly.
The first type of reactions was the halogen (Cl or Br) insertion on the substance phenol ring. The treatment of (1) with N-chlorosucccinamide (NCS) in carbon tetrachloride solution in the presence of triethylamine (TEA) resulted in high yield (91%) of its monochloroderivative (2).
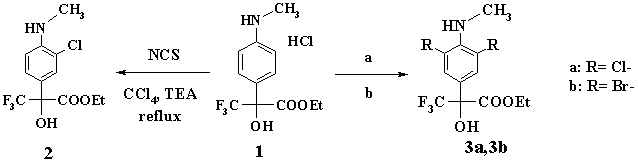
(Procedure (a): NCS, CCl4, boiling. Procedure (b): Br2, AcOH, 0-5В°C.)
To prepare 3,5-dichloroderivative (3a) threefold NCS excess was used in the process. The yield of (3a) was 72%. The bromation of (1) with molecular bromine in acetic acid solution resulted in good yield (60%) of 3,5-dibromoderivative (3b).
The second type of transformation reactions resulted in aminohydroxy acid (4) thanks to saponification of (1) in water-dioxane solution in the presence of surplus NaOH. The yield of (4) was 84%.
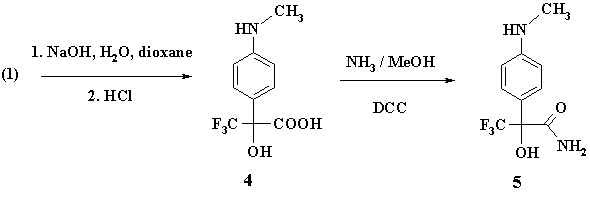
Further the acid (4) was converted to amide (5) by its treatment with methanol solution of ammonia (~15% mass) in the presence of dicyclohexylcarbodiimide (DCC); the yield of amide (5) was 50%. Amide (5) was also produced by ammonolysis of (1) with water solution of ammonia with even higher yield (73%).
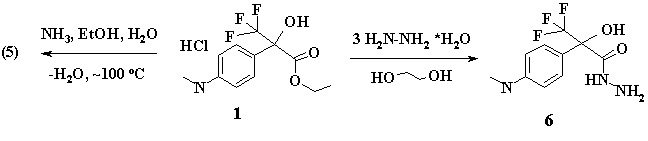
Boiling of (1) with threefold excess of hydrazine hydrate resulted in carbohydrazide (6), its yield was 62%.
The third type of transformations was aimed to prepare alkylderivatives by the hydroxyl group of (1), or to reduce (C-OH) to form (C-H) bond. To this purpose (1) was treated with thionyl chloride in chloroform solution to give (7). The resulting substance (7) in the further transformations was used without isolation. In the following reactions the presence of initial (1) was not revealed, thus evidencing its exhaustive conversion. The formation of (7) was confirmed by mass-spectrometry. It should be noted that chlorinated derivative (7) is not stable, and in the presence of water it is easily hydrolyzed resulting in chlorine split-off and the original substance (1) formation.
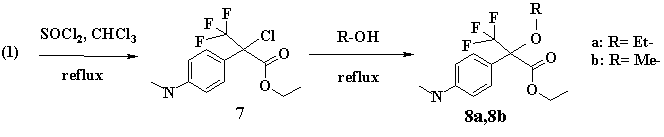
Thus, boiling of chlorinated derivative (7) in ethanol resulted in floroksan ethoxyderivative (8a) with good yield (72%). The use of methanol led to the formation of its methoxyderivative (8b) with a little higher yield (75%).
The easiness of chlorine nucleophilic substitution at this position must be apparently due to the influence of neighboring acceptor groups (trimethyl- and ester- groups) that increase positive charge on the carbon atom of benzyl. The phenyl ring favors stabilization of the carbocation due to the formation of parachinoid structure. The reduction of (C-Cl) bond in (7) was carried out by activated zinc in glacial acetic acid solution [4]. The substance (9) yield, as recalculated on 2-stage basis, was 78%.
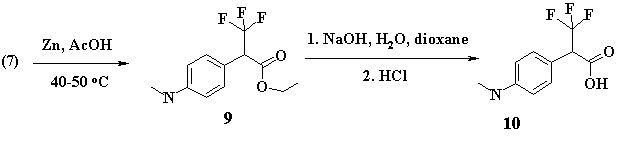
Alkaline-based saponification of ester (9) in water-dioxane solution, in the presence of sodium hydroxide, resulted in aminoacid (10) with good yield (67%), as recalculated on 2-stage basis.
The approaches to modification, and search for novel "floroksan" derivatives are to be designed after biological testing of newly prepared substances.
Experimental
Both structure and purity of the products were confirmed using thin-layer chromatography (Kieselgel 60 F254 plates), NMR-spectroscopy (Avance 300 for 1H, 19F), and chromato-mass-spectrometry (Finnigan Polaris Q/Trace GC ultra, ionic trap, EU 70 eV. Column: RTX-5ms, phase type: 5% phenylpolysilphenylene-siloxane).
Ethyl ester of 2-(3-chloro-4-methylamino-phenyl)-3,3,3-trifluoro-2-hydroxy propionic acid (2): 0.33g (0.0033mole) of TEA and 0.43g (0.0032mole) of NCS were added to 1g (0.0032mole) of (1) dissolved in 50 ml of CCl4, and boiled during 2 hours with a reflux condenser, the контроль зa ходом reaction was TLC-controlled till complete disappearance of original (1), eluent flow was: chloroform/ethylacetate with ratio 95/5. Pyrrolidinedione that had precipitated after the reaction completion was filtered, and the solution was boiled down on a rotor evaporator under reduced pressure, the boiled down mass was purified by chromatography on 150g of SiO2, eluent flow was: chloroform/petroleum-ether at ratio 9/1, Rf=0.75. It resulted in 0.9g of substance (2), yield 91%. Slow crystallizing light-yellow oil.
1H NMR spectrum: dmso-d6; 1.2 ppm: 3H-t, j=7Hz, CH3; 2.76 ppm: 3H-d, j= 7Hz, N-CH3; 4.24 ppm: 2H-q, j= 7Hz, CH2; 5.8 ppm: 1H-m-widened, N-H; 6.65 ppm: 1H-d, j= 7.5Hz, Aryl; 7.28 ppm: 1H-dd, Jo= 7.5Hz, Jm= 0.13Hz, Aryl; 7.38 ppm: 1H-d, jm= 0.13Hz, Ar.; 7.65 ppm: 1H-c, OH.
NMR-19F Spectrum: dmso-d6; 2.48 ppm singlet, CF3.
Mass-spectrum: m/z 311 [M]+ (33.2); 272 [C12H12ClFNO3]+ (3.2); 238 [C9H8ClF3NO]+, (100); 168 [M-CF3-CO2-H2O]+ (88.3); 141 [M-C5H5F3NO2]+ (23.2).
Ethyl ester of 2-(3,5-dichloro-4-methylamino-phenyl)-3,3,3-trifluoro-2-hydroxypropionic acid (3a): 1.3g (0.0096mole) of NCS were added to 1g (0.0032mole) of (1) dissolved in CCl4, and boiled during 4 hours. The reaction was TLC-controlled, eluent flow was: chloroform/ petroleum-ether with ratio 9/1, Rf= 0.7. The isolation and purification procedure for (3a) was similar to that for substance (2). It resulted in 0.8g, with yield 72%. Slow crystallizing light-yellow oil.
NMR-1H Spectrum: dmso-d6; 1.22 ppm: 3H-t, j=7Hz, CH3; 3.0 ppm: 3H-c, N-CH3; 4.27 ppm: 2H-q, j=7Hz, CH2; 5.39 ppm: 1H-widened, N-H; 7.39 ppm: 2H-c, Aryl; 7.96 ppm: 1H-c, -OH.
NMR-19F Spectrum: dmso-d6; 2.51 ppm singlet, CF3.
Mass-spectrum: m/z 345 [Рњ]+ (30.2); 311 [C11H9Cl2F2NO3]+ (18.1); 272 [M-C3H5O2]+ (100).
Ethyl ester of 2-(3,5-Dibromo-4-methylamino-phenyl)-3,3,3-trifluoro-2-hydroxy propionic acid (3b): To 1g (0.0032mole) of (1) dissolved in 20ml of glacial acetic acid at 0-5В°C 1.04g (0.0065mole) of bromine was added. The reaction was TLC-controlled till complete disappearance of original (1), eluent flow was: chloroform/petroleum-ether at ratio 9/1, Rf=0.8. After that the reaction blend was boiled down at reduced pressure and the residuum was moved into 50ml of water. The water solution was brought to СЂH=7 by adding solid NaHCO3 by small portions. Precipitated crystals were filtered, washed with water and did not require further purification. The yield of 3,5-dibromo derivative (3b) was 0.83g, 60%. White crystal substance, Рўmelt=135-138 В°C.
NMR-1H Spectrum: dmso-d6; 1.22ppm: 3H-t, j=7Hz; 2.95ppm: 3H-d, j=7Hz, N-Me; 4.27ppm: 2H-q, j=7Hz, CH2; 5.00 ppm: 2H-q widened, j=7Hz, N-H; 7.62 ppm: 2H-s, widened, Ar; 8.00 ppm: 1H-s, widened, -OH.
NMR-19F Spectrum: dmso-d6; 2.56 ppm singlet widened, -CF3.
Mass-spectrum: m/z 433 [Рњ]+ (100.00); 362 [C9H8Br2F3NO]+ (62.5); 292 [C11H12Br2NO3]+ (82).
3,3,3-Trifluoro–2–hydroxy–2–(4-methylamino-phenyl)-propionic acid (4): 0.3g (0.007mole) (1.1 equivalent) of NaOH in 5ml of water was added to 20ml of water-dioxane (1/1 ratio) solution of 2g (0.0064mole) of (1) and boiled during two hours. After that the reaction mass was cooled and dioxane was boiled down on a rotor evaporator under reduced pressure. The resulted water-soluble mass was cooled to 0-5°C and acidified with muriatic acid (18%) to neutral рH, sedimented acid (4) was filtered and washed on a filter with distilled water. The yield of (4) was 1.3g, 84%. Light-grey crystal substance, Тmelt=162-167 °C, with decomposition.
NMR-1H Spectrum: dmso-d6; 2,6 ppm: 3H-d, N-CH3; 6.54 ppm: 2H-d, j= 7.5Hz, Aryl; 7.3 ppm: 2H-d, j= 7.5Hz, Aryl.
NMR-19F Spectrum: dmso-d6; 2.52 ppm singlet, -CF3.
Mass-spectrum: m/z 249[M]+ (9.1); 231 [M-H2O]+ (4.9); 204 [M-CHO2]+ (80.7); 134 [M-CHO2-CF3-H]+ (100); 107 [C7H9N]+ (37.5).
3,3,3-Trifluoro-2-hydroxy-2-(4-methylamino-phenyl)-propionamide (5): Procedure 1. 0.41g (0.002mole) of DCC at room temperature was added to 0.5Рі (0.002mole) of (4) dissolved in 15ml of methanol (with ~15% mass of ammonia). Next day the reaction mass was boiled down, solid residuum was purified by chromatography on 150g of SiO2, eluent flow was: chloroform/ethylacetate at ratio 8/2, Rf=0.25. The yield of amide (5) was 0.25g, 50%.
Procedure 2. Amide (5) was produced by boiling with an efficient reflux condenser during 3 hours of 10ml of alcohol and 15ml of water-ammonia solution that contained 1g (0.0032mole) of (1). The reaction was TLC-controlled, eluent flow was: chloroform/ethylacetate at ratio 8/2, Rf=0.25. On completion of the reaction water-alcohol solution was half-boiled down, its cooling resulted in precipitation of crystal (5). The yield of amide (5) was 1.15g, 73%. Light-grey crystal substance, Рўmelt=183-188 В°C.
NMR-1H Spectrum: dmso-d6; 2.67 ppm: 3H-s, N-CH3; 5.75 ppm: 1H, уширен, NH; 6.49 ppm: 2H-d, j=8Hz, Aryl; 7.14 ppm 1H-s, widened, -OH; 7.32 ppm: 2H-d, j=8Hz, Aryl, 7.52 ppm: 2H-s, widened, -NH2.
NMR-19F Spectrum: dmso-d6; 4.15 ppm singlet, -CF3.
Mass-spectrum: m/z 248 [Рњ]+ (18.5); 204 [C9H9F3NO]+ (87.5).
Hydrazide of 3,3,3-trifluoro-2-hydroxy-2-(4-methylaminophenyl) propionic acid (6): 0.25g (0.0048mole) of hydrazine monohydrate was added to 0.5g (0.0016mole) of (1) dissolved in 10ml of ethylene glycol and boiled during three hours with a reflux condenser. Then the reaction mass was boiled down on a rotor evaporator under reduced pressure, the rest of mass moved into 20ml of water, and precipitated crystals were filtered and washed with water, no further purification was required. The yield of substance (6) was 0.42g, 62%. White crystal substance, Рўmelt=151-155 В°C. The reaction was TLC-controlled, eluent flow was chloroform/ethylacetate at ratio 85/15, Rf=0.15.
NMR-1H Spectrum: dmso-d6; 2.64 ppm: 3H-d, j=5,5Hz, CH3; 4.32 ppm: 2H-s, NH2; 5.8 ppm: 1H-d, j=4Hz, NH; 6.49 ppm: 2H-d, j=9Hz, Aryl;; 7.2 ppm: 1H-s, OH; 7.34 ppm: 2H-d, j=9Hz, Aryl.
NMR-19F Spectrum: dmso-d6; 3.96 ppm singlet, CF3.
Mass-spectrum: m/z 263 [Рњ]+ (8.4); 204 [C9H9F3NO]+ (61.2); 176 [C8H5F3O]+ (2.5).
Ethyl ester of 2-chloro-3,3,3-trifluoro-2-(4-methylamino-phenyl)-propionic acid (7): 1.52g (0.0128mole) of freshly distilled thionyl chloride was dropwise added to 20ml of 2g (0.0064mole) of substance (1) dissolved in cooled to 0-5В°C chloroform, that contained also catalytic amount of pyridine (50mg). The reaction mass was boiled during 4 hours with a reflux condenser and calcium chloride tube. The reaction was TLC-controlled, eluent flow was: dichloromethane/petroleum-ether (40-70) at ratio 1/1, Rf=0.5. After that the reaction mass was boiled down on a rotor evaporator under reduced pressure till thick residuum formation, and re-boiled down with chloroform in order to remove any trace of thionyl chloride.
Mass-spectrum: m/z 295 [Рњ]+ (15.6); 260 [M-Cl]+ (42.6); 222 [M-C3H5O2]+ (100); 188 [M-C3H4O2-Cl]+ (76.9).
Ethyl ester of 2-ethoxy-3,3,3-trifluoro-2-(4-methylamino-phenyl)-propionic acid (8a): 20ml of absolute ethanol was added to prepared substance (7) (0.0064mole) and boiled with a reflux condenser during 3 hours. The reaction was TLC-controlled, eluent flow was: dichloromethane/petroleum-ether (40-70) at ratio 1/1, Rf=0.75. Reaction mass was boiled down, and solid residuum was purified by chromatography on 150g of SiO2, eluent flow was: dichloromethane/petroleum-ether (40-70) at ratio 1/1, Rf=0.75. The yield of (8a) was 1.4g, 72% as recalculated to two steps. Light-yellow oil.
NMR-1H Spectrum: CDCl3: 1.28 ppm: 6H-t, j=5.5 Hz, 2-CH3; 2.84 ppm: 3H-s, N-Me; 3.54-3.79 ppm: 2H-m, j=7 Hz, -O-CH2-; 4.34 ppm: 2H-m, j=7 Hz, -O-CH2- ester; 6.56 ppm: 2H-d, j=10 Hz, Aryl; 7.35 ppm: 2H-d, j=10 Hz, Aryl.
NMR-19F Spectrum: CDCl3: 5.45 ppm: s, -CF3.
Chromato-mass spectrum: purity ≥ 95%, retention time in a capillary column was 21.03 min, m/z 305 [Рњ]+ (6.3); 232 [C12H13F3NOВ3]+ (44); 134 [C8H8NO]+ (100); 107 [C7H9N]+ (20.8).
Ethyl ester of 3,3,3-trifluoro-2-methoxy-2-(4-methylamino-phenyl)-propionic acid (8b): Substance was produced similar to (8a). The yield of substance (8b) was 1.35g, 75% as recalculated to two-steps. Light-yellow oil, Rf=0.75.
NMR-1H Spectrum: CDCl3: 1.33 ppm: 3H-t, j= 10Hz, -Me; 2.84 ppm: 3H-s, -N-Me; 3.48 ppm: 3H-d, j=1 Hz, -O-Me; 4.36 ppm: 2H-d, j= 10 Hz, -CH2-; 6.57 ppm: 2H-d, j= 10 Hz, Aryl; 7.27 ppm: 2H-d, j= 10 Hz, Aryl.
NMR-19F Spectrum: CDCl3: -72.1 ppm: s, -CF3.
Chromato-mass spectrum: purity ≥ 95%, retention time in capillary column was 20.9 min, 291 [М]+ (21.4); 260 [M-CH3O]+ (3.2); 218 [M-C3H5O2]+ (100); 134 [C8H8NO]+ (58.0).
Ethyl ester of 3,3,3-trifluoro-2-(4-methylamino-phenyl)-propionic acid (9): 20 ml of glacial acetic acid and 3g of activated zinc dust under permanent stirring were added to the prepared substance (7) (0.0064mole), keeping the temperature 40-50 В°C during three hours. The reaction was TLC-controlled, eluent flow was: chloroform/ethylacetate at ratio 97/3, Rf=0.7. After that the acetic acid solution was boiled down, and the rest of mass moved into 50ml of water. The water solution СЂH was made 8 by adding NaHCO3 in small portions, and the water solution was extracted with ether. The ether extract (3 times by 50ml) was put together, dried with waterless sodium sulfate and boiled down on a rotor evaporator under reduced pressure. The residuum was purified with chromatography on 200ml of SiO2, eluent flow was: chloroform/ethyl acetate at ratio 95/5. The yield was 1.3g, 78%. Light-yellow oil.
NMR-1H Spectrum: CDCl3: 1.25 ppm: 3H-t, j= 10Hz, -Me; 2.83 ppm: 3H-s, N-Me; 4.16- 4.25 ppm: 2H-q, j=10 Hz, -CH2-, 1H-m, -CH*-; 6.57 ppm: 2H-d, j= 10 Hz, Aryl; 7.25 ppm: 2H-d, j= 10 Hz, Aryl.
NMR-19F Spectrum: CDCl3: 9.45 ppm: d, j= 10 Hz, -CF3.
Chromato-mass spectrum: purity ≥ 97%, retention time in capillary column 19.01 min, m/z 261 [М]+ (18.6); 188 [M-C3H5O2]+ (100); 138 [C8H9FN]+ (48.8).
3,3,3-Trifluoro-2-(4-methylamino-phenyl)-propionic acid (10): The preparation procedure was similar to that for (4). Light-grey crystal substance, Рўmelt= 142-147 В°C with decomposition. The yield was 1.11g, 67% as recalculated to two steps.
NMR-1H Spectrum:; 2.77 ppm: 3H-s, N-Me; 4.35 ppm: 1H-m, -CH*-; 6.77 ppm: 2H-d, j= 10 Hz, Aryl; 7.35 ppm: 2H-d, j= 10 Hz, Aryl.
NMR-19F Spectrum: dmso-d6: 9.65 ppm: d, j= 10 Hz, -CF3.
References
- Regulatory rosta rastenii v sel'skom hozyaistve (Plant growth regulators in agriculture)./ Ed. G. Tukei, Moscow: Izd. Inostr. lit., 1958. – 387 p.
- N.D. Chkanikov, V.D. Sviridov, A.A. Kadyrov, Yu.Ya. Spiridonov. Seed treating composition with growth regulation effect. RU Patent No. 2369094 (2009), Bull. Izobr. No. 28 (2009).
- G.S. Muromtsev, D.I. Chkanikov, O.N. Kulaeva, K.Z. Gamburg. Osnovy khimicheskoi regulyatsii rosta i produktivnosti rastenii (Principles of chemical regulation of plant growth and productivity), Moscow: Agropromizdat, 1987. – 383 p.
- V.I. Dyachenko, A.S. Peregudov, N.D. Chkanikov. Trifluoromethyl-containing N-acylmethylenequinone imines as novel highly electrophilic agents. J. Fluorine Chem., 2007, 128 (7), 868 - 878.
Recommended for publication by Prof. N.D. Chkanikov
Fluorine Notes, 2014, 92, 3-4
