Received: December, 2012
Fluorine Notes, 2013, 89, 7-8
Structural Properties of Theoretical Studies of Vanadium(III) fluoride and Molybdenum(III) fluoride Compounds*
Ali Rahmani
Department of Chemistry, Faculty of Science, Azad University unit Of Takestan , Takestan, Iran.
Fax: (+98) 281-3780040, e-mail: Ali_Rahmani870@yahoo.com
Abstract: In this theoretical study we used density functional theory to calculate the molecular structures of Vanadium(III) fluoride and Molybdenum(III) fluoride Compounds VF3 and MoF3. The molecular geometry, vibrational frequencies, energies and natural bond orbital (NBO) in the ground state are calculated by using the DFT (B3LYP) methods with LANL2DZ. The T.S guesses were generated by the linear synchronous transit method, at the DFT implemented on Gaussian 09 program. The geometries and normal modes of vibrations obtained from B3LYP calculations are in good agreement with the experimentally observed data.
Keywords: Theoretical studies, Vanadium(III) fluoride and Molybdenum(III) Compounds, Gaussian 09, VF3 and MoF3
1. Introduction
The unique properties of halide impart an unusual reactivity to the fluoride bonds which can be exploited in preparative inorganic chemistry or in catalysis. Investigation of the structures and properties of these compounds and their similarities are interested.
Flourine compounds have many used in theoretical and industrial. Many different data have been found about the structural properties of flour compounds, but they are insufficient and opposing in somewhere. Due to the extreme electronegativity of fluorine, perfluorinated compounds have unique physical properties which are useful in organic synthesis and separation methods such as solid phase extraction. Fluorine compounds have many used in theoretical and industrial. Many different data have been found about the structural properties of fluoro compounds, but they are insufficient and opposing in somewhere. fluoro compounds have many used in theoretical and industrial. Many different data have been found about the structural properties of fluoro compounds, but they are insufficient and opposing in somewhere. Two primitive synthesized Vanadium(III) fluoride and Molybdenum(III) compounds are VF3 and MoF3, which used for structural chemistry studies and organic synthesis.
Theoretical calculations have been used for extraction of structural and electronically data of many compounds especially flour compounds. We applied the DFT method to optimize and calculate molecular data of synthesized compounds. The calculation was done by using the Gaussian 09 programs AD (1993), Becke For DFT, Becke's three-parameter exchange functional CT (1988). Lee was used in combination with the Lee-Yang-Parr correlation functional (B3LYP) with LANL2DZ basis set. Seppelt synthesized Vanadium(III) fluoride and Molybdenum (III) Compounds and in this paper we investigated other properties of them [1-11].
2. Material and methods
2.1 Computational methods
All computational are carried out using Gaussian 09 program [12, 13] which combines the exact Hartree-Fock exchange with Becke,s and uses the Lee-Yang-Parr correlation function in order to include the most important correlation effects. The structures of the molecules were completely optimized without any symmetry in all the levels. The optimized structural parameters were used in the vibrational frequency calculations at the DFT levels to characterize all stationary points as minima. Infrared intensities (int) in Kilometer per mole of all compounds were performed at the same level on the respective fully optimized geometries. These compounds and their data are in accordance with recent works on the formation of four coordinate intermediates LANL2DZ.
3. Results
Vanadium(III) fluoride and Molybdenum(III) compounds are VF3 and MoF3, were studied and geometry optimizations were performed at the B3LYP / LANL2DZ level and are shown in Figure. 1. The VF3, in which F (2) is bonded to the V (1) atom, has linear F(2)-V(1) structure with bond length 2.59 Å. F(3) is bonded to the V(1) atom, has a linear F(3)-V(1) structure with bond length 2.61 Å. The MoF3, in which F(2) is bonded to the Mo(1) atom, have a linear F(2)- Mo(1) structure with bond length 2.06 Å. F(4) is bonded to the V(1) atom, have a linear F(4)-V(1) structure with bond length 2.29 Å. Selected bond distances are illustrated in Figure. 1. Selected angles are reported in Table 1.
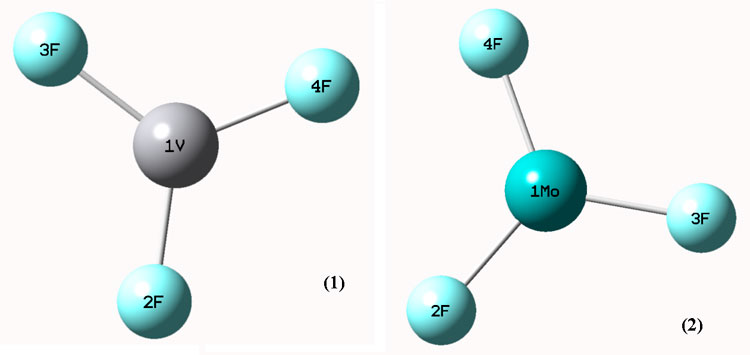
Figure.1. Optimized geometries of VF3 and Mo3 at B3LYP/LANL2DZ level of theory
Table 1. Geometrical parameters optimized for (1) VF3, (2) Mo3 some selected bond lengths (Å) and angles (°).
Both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the main orbitals that take part in chemical stability. The HOMO represents the ability to donate an electron, LUMO as an electron acceptor represents the ability to obtain an electron the HOMO and LUMO energy calculated by B3LYP at LANL2DZ method (Figures. 2)'. This electronic absorption corresponds to the transition from the ground to the first excited state and is mainly described by one electron excitation from the highest occupied molecular or orbital (LUMO).
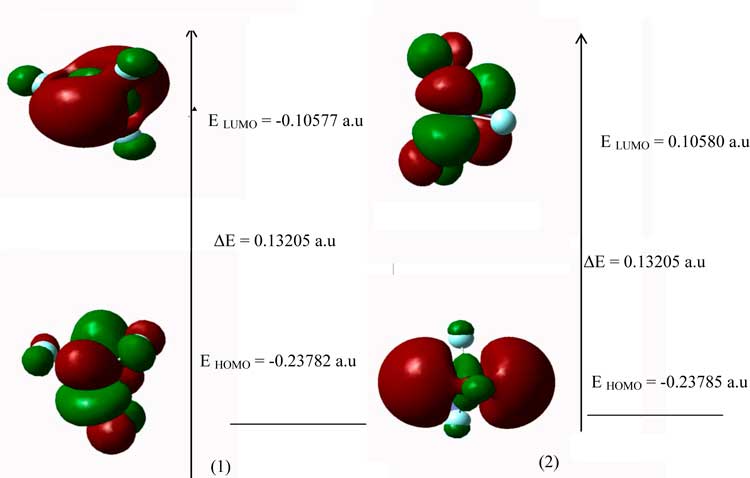
Figure2. The atomic orbital of the frontier molecular orbital for (1) VF3, (2) Mo3 at B3LYP/LanL2DZ level of theory.
Atomic charges and bond orders are significant parameters for our investigation. These quantities are derived from the NBO population analysis. The NBO method is preferred to Mulliken charges, because the former provides an orbital picture that is closer to the classical Lewis structure. The NBO analysis involving atomic charges, bond orders as well as hybridizations of selected bonds are calculated at B3LYP/LANL2DZ level.
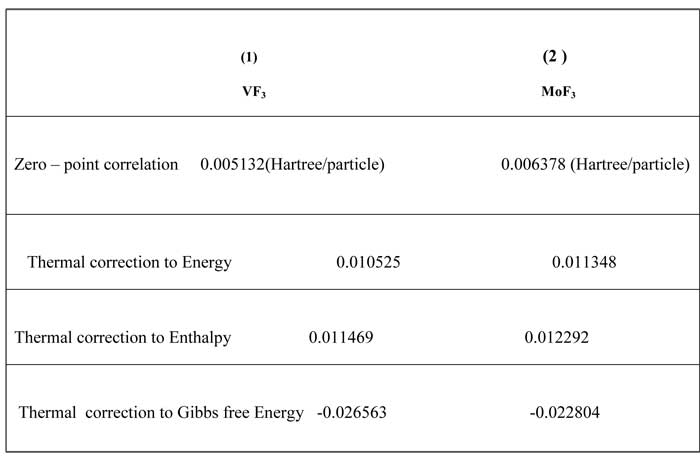
The NBO calculated hybridization for flour carbon compounds, VF3 and MoF3 shows that all of compounds have SPXhybridization and non planar configurations (Table 2). Second order perturbation theory analysis of Fock matrix in NBO basis for flour carbon compounds, VF3 and MoF3 is shown in( Table 3 ).Some thermodynamic parameters Frequencies for (1) VF3, (2) MoF3 Zero-point Energy, correction Energy, Enthalpy lengths, Gibbs free Energy are calculated and confirmed with other published theoretical data. ( Table 4).
Table2. The NBO Calculated Hybridizations for (1) VF3, (2) Mo3 at the B3LYP/LanL2DZ.
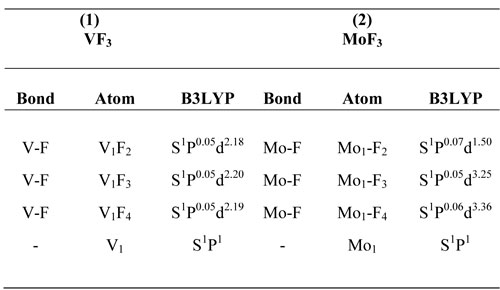
Table3. Second order perturbation theory analysis of Fock matrix in NBO basis
for (1) VF3, (2) Mo3
a means energy of hyper conjugative
interaction (stabilization energy);
b Energy difference between donor and
acceptor i and j NBO orbital's;
c F(i, j) is the Fock matrix element between i and j NBO orbital's.
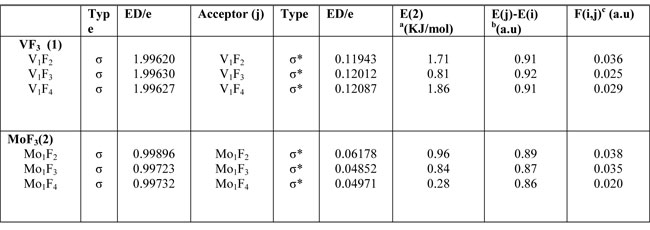
Table4. Some thermodynamic parameters Frequencies for (1) VF3, (2) Mo3 Zero-point Energy, correction Energy, Enthalpy lengths, Gibbs free Energy.
These molecules have distorded C3v symmetries respectively and trigonal and square pyramidal shapes.
4. Discussion
Density functional theorymethods were employed to determine the optimized structures of VF3 and MoF3. Initial calculations were performed at the DFT level and split - valence plus polarization LANL2DZ basis sets were used. Local minima were obtained by full geometrical optimization have all positive frequencies. All calculations were carried out using the computer program GAUSSIAN 09.
4.1 NBO study on structures
Natural Bond Orbital's (NBOs) are localized few-center orbital's that describe the Lewis-like molecular bonding pattern of electron pairs in optimally compact form. More precisely, NBOs are an orthonormal set of localized "maximum occupancy" orbital's whose leading N/2 members (or N members in the open-shell case) give the most accurate possible Lewis-like description of the total N-electron density. This analysis is carried out by examining all possible interactions between "filled" (donor) Lewis-type NBOs and "empty" (acceptor) non-Lewis NBOs, and estimating their energetic importance by 2nd-order perturbation theory. Since these interactions lead to donation of occupancy from the localized NBOs of the idealized Lewis structure into the empty non-Lewis orbitals (and thus, to departures from the idealized Lewis structure description), they are referred to as "delocalization" corrections to the zeroth-order natural Lewis structure. Natural charges have been computed using natural bond orbital (NBO) module implemented in Gaussian09. The NBO Calculated Hybridizations are significant parameters for our investigation. These quantities are derived from the NBO population analysis. The former provides an orbital picture that is closer to the classical Lewis structure.
4.2 Frontier molecular orbital
Both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the main orbital take part in chemical stability. The HOMO represents the ability to donate an electron, LUMO as an electron acceptor represents the ability to obtain an electron. The HOMO and LUMO energy were calculated by B3LYP/LANL2DZ method. This electronic absorption corresponds to the transition from the ground to the first excited state and is mainly described by one electron excitation from the highest occupied molecular or orbital (LUMO). Therefore, while the energy of the HOMO is directly related to the ionization potential, LUMO energy is directly related to the electron affinity. Energy difference between HOMO and LUMO orbital is called as energy gap that is an important stability for structures.
Conclusion
In this theoretical study we used density functional theory to calculate the molecular structures of the flour carbon compounds, VF3 and MoF3. The molecular geometry, vibrational frequencies, energies and natural bond orbital (NBO) in the ground state are calculated by using the DFT (B3LYP) methods with LANL2DZ.
Acknowledgements. We gratefully acknowledge the financial support from the Research Council of Takestan Islamic Azad University.
References
- K.Seppelt, W. Sundermeyer, N-Halogensulfinylamine.Naturwissenschaften, 1969, 56: 281-282.
- Arno Schmuck, Konrad Seppelt, Sulfur Pentafluoride Cyanate F5S-O-C≡N. Angewandte Chemie International Edition in English, February 1987, Volume 26, Issue 2, pages 134-135,
- K.Seppelt, W. Sundermeyer, EineneueMethodezurHerstellung von Schwefeloxidtetrafluorid, 1971, ZAAC, 386:229-231.
- K. Seppelt, H. H.Eysel, Schwingungsspektren und Kraftkonstanten des Tetrakistrimethylsilylhydrazin, Anorg. Allg. Chem, 1971, 384:147-154.
- Bruce Sutherland, R., M. Ho.Douglas, John Huffman, C. Caulton, Kenneth G, Heterometallic Polyhydride Raft Formation: A Comparison of Syntheses using Alkoxides of Copper and Gold. AngewandteChemie International Edition in English, 1987, 26:135-137.
- K.Seppelt, W.Sundermeyer, Über N-Halogenimidoschwefeldifluoride und N,N'-Dihalogenschwefeldiimide. Angew.Chem, 1969, 81:785-786.
- K.Seppelt, W.Sundermeyer, NeueImidoschwefeloxiddifluoride. Angew. Chem, 1970, 82:931-955.
- K.Seppelt, H.Oberhammer, Pentafluoroseleniumcyanate, F5SeOC.tplbond.N. Inorg.Chem, 1985, 24: 1227-1229.
- Becke, A.D. 1993. Density-functional thermochemistry. III. The role of exact exchange. J.Chem. Phys, 98: 5648-5652.
- Sundaraganesan, N.,S. Ilakiamani,B.Dominic Joshua, Vibrational Spectroscopyinvestigation using ab initio and density functional theory analysis on the structure of 3, 4-dimethylbenzaldehyde.Spectrochimica Acta Part A, 2007, 68:680-687.
- K.Seppelt, W.Sundermeyer, 1970. NotizüberMetalltris(trimethylsilyl)hydrazide, Chem.Ber, 103: 3939-3941.
- A. Roland, K.Seppelt, W. Sundermeyer, 19F-Kernresonanzen an Imidoschwefeloxiddifluoriden, Z. anorg.allg. Chem, (1972), 393, 149-151.
- K.Seppelt, Arsenpentachlorid, Angew. Chem. (1976), 88, 410-411.
*retained original spelling
Recommended for publication by S. Igoumnov
Fluorine Notes, 2013, 89, 7-8
