Received: July, 2011
Fluorine Notes, 2013, 87, 1-2
Manifestation of Complicated Structure of Potential Energy Surfaces in Spectral and Chemical Properties
of Haloarene Radical Ions
Shchegoleva L.N., Beregovaya I.V.
N.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry of the Siberian Branch of Russian Academy
of Sciences, Novosibirsk, Russia
E-mail: sln@nioch.nsc.ru,
ivb@nioch.nsc.ru
Abstract: This article presents results of systematic quantum chemical studies on the electron and spatial structure of radical ions of polyfluoro- and some chlorine containing compounds of the benzene row. The reasons and regularities of structure distortions in these radical ions were analyzed and their manifestations in the ESR spectra were discussed. The relationship between the potential energy surface structure of haloarene radical anions and their ability to halide-ion elimination has been investigated and the reaction mechanism has been studied in details.
Keywords: Fluorinated arenas, radical ions, quantum chemical calculations, structure distortions, potential energy surfaces, conical intersections, ESR, reactivity, halide-ion elimination.
Table of Contents
I. Introduction
II. Evolution of Ideas on Structure of Fluoroarene Radicals Ions
III. Methodology of Potential Energy Surface Calculations
IV. Adiabatic Potential Energy Surfaces of the Benzene and Hexafluorobenzene Radical Ions
V. Potential Energy Surfaces of Radical Ions of Partially Fluorinated Benzenes
VI. Manifestation of Structure Flexibility in the ESR Spectra of Polyfluoroarene Radical Ions
VII. Structural Distortions and Monomolecular Cleavage of Haloarene Radical Anions
I. Introduction
In modern organic chemistry more and more attention is being paid to the detailed studying of intermediate particles. Their identification, knowledge of their structure and properties allow us to judge on elementary stages and patterns of chemical processes. Many chemical and electrochemical transformations of aromatic and unsaturated compounds as well as biological processes involve intermediate forming of radical ions. Radical cations are detected under the conditions of electrophilic substitution reactions, radical anions come as the key intermediates for the SRN1 nucleophilic substitution,2 they participate in the mechanisms of the Griniar reagents formation3 and DNA damage4.
The progress in studies on the radical ion intermediates is connected to a great extent with the improvement of experimental physical methods particularly ESR. The methods of generation of short-lived radical ions and their stabilization in matrices at low temperature and also the method of optical detection a radical ion pairs in solutions (OD ESR)5 play a large part as well. Quantum chemical studies are of no less significance. Modern day development of ab initio methods of quantum chemistry and their program implementation presents opportunities for both theoretical interpretation of experimental results and obtaining of independent information about structure and properties of short-lived intermediates and transition states of chemical reaction, which is hard-to-reach with experimental methods.
II. Evolution of Ideas on Structure of the Fluoroarene Radicals Ions
Particular interest to structure and stability of radical ions of fluoroaromatic compounds is due to their considerable role in the fluoroarenes chemistry, which substantially differs from the chemistry of non-fluorinated analogues and results in obtaining new products of practical value – biologically active compounds, medicines, polymer materials etc.7-12
Radical cations of a wide series of polyfluroaromatic compounds have been generated for the first time in the late 60-s in the Halogen Compounds Laboratory of Novosibirsk Institute of Organic Chemistry of Siberian Branch of Russian Academy of Science.13,14 That particles proved to be stable enough to register their ESR, UV and IR spectra. Interpretation of the spectral data has required to attract quantum chemical calculations, and it was a beginning for the regular theoretical studies of polyfluoroarene radical ions.
First calculations have been done in π-electronic approximation using the Hückel, McLachlan and Pariser–Parr–Pople methods and geometrical parameters of neutral precursors. And even these simple calculations allowed interpreting the rather complicated ESR spectra of heptafluoronaphthalene radical cations with seven non-equivalent 19F nuclei.15 Later the methods treating all valence electrons (CNDO/2, CNDO/S, INDO) became available. The results obtained during interpretation of hfc constants and absorption spectra of polyfluoroaromatic radical cations 16,17 fully corresponded to the ideas regarding radical ions of aromatic molecules as planar π-radicals 18,19 that had formed in the world literature. Hfc constants in the ESR spectra of such radicals are caused only by spin polarization and as a rule they do not exceed 10 G for 1H nuclei and 50 G for 19F nuclei.
Radical anions of polyfluoroaromatic molecules are far less stable than respective cations. For the first time Williams and co-workers 20,21 managed to register them in the 70-s owing to the development of matrix isolation methods at low temperatures. Unexpectedly large values of hfc constants aF (~100-200 G), found for radical anions of hexa- and pentafluorobenzenes obviously did not correspond to the idea of planar π-radical structure. The problem of origin of abnormally great aF values in radical anions of fluorinated benzenes has been being intensively discussed in literature for almost 10 years, at that two main hypotheses have been mentioned – planar σ-radicals and distorted non-planar structures. First of them is based on the proposition of crossing the π and σ states levels at substitution of hydrogen atoms by fluorine atoms (according to the data of photoelectronic spectroscopy it is known,22 that under the fluorine atoms accumulation σ-levels lower faster, than π ones). However, according to the data of electron capture spectroscopy23 in the gas phase polyfluorobenzene molecules capture electron onto π level. Besides that, the model of planar σ-radical does not explain the observed particularity of hfc anisotropy (A|F > A⊥F),24 which lies in the grounds of the second hypothesis. The data of different experiments contradicted each other and quantum chemical calculations were too imperfect to make a definite conclusion.
Both experimental and theoretical works regarding studying of polyfluoroaromatic radical anions were being carried out in Novosibirsk Institute of Organic Chemistry in cooperation with Institute of Chemical Kinetics and Combustion SBRAS. We managed to solve the problem of those particles structure by means of quantum chemical (INDO) analysis of structural dependence of hfc constants within the model based on vibronic (pseudo-Jahn-Teller) interaction of the ground ПЂ and excited Пѓ states of radical anion.25 Vibronic interaction results in the out-of-plane structure distortions with F atoms deviation from the ring plane, at that symmetry of distorting (active) coordinates is defined by the symmetry of interacting states. The reason for this model were the INDO calculations data on closing (but not crossing) of ПЂ and Пѓ levels at substitution of H atoms by F. Later these data have been confirmed by ab initio calculations (Fig. 1). The scheme of levels shown in Fig. 1 explains not only the origin of out-of-plane deformations in the hexafluorobenzene anion, but also the planar structure of the respective cation, for which the substitution of H atoms by F leads to the opposite effect.
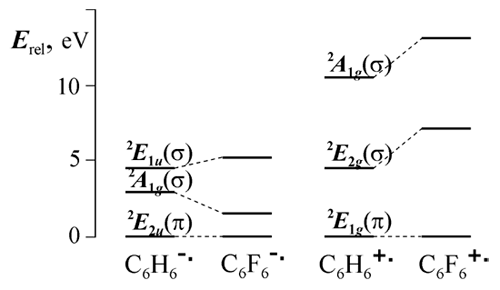
Figure 1. Relative energy levels of the lower electronic states of the benzene and hexafluorobenzene radical ions (ROHF/6-31G*).
The offered model allowed the regularities of the out-of-plane distortions arising to be revealed and explained the ESR data for radical anions of a wide range of fluorine containing benzene derivatives, among which there are both planar ПЂ- and Пѓ-radicals and distorted non-planar pseudo-ПЂ- and pseudo-Пѓ-structures which single occupied MOs (SOMOs) are linear combinations of ПЂ and Пѓ orbitals. The presence of direct contribution of the fluorine sВ AРћ in SOMO explains the origin of unusually large values of hfc constants with 19F nuclei at the distorting of ПЂ-radical planar structures.
The reason for out-of-plane distortions is an interaction of electronic states of different symmetry types – π (symmetric) and σ (antisymmetric) – in regards to the plane of conjugated system. Further studies within framework of vibronic approach showed, that in-plane structure distortions caused by interaction between the states of the same symmetry type (π and π or σ and σ) are also possible in radical ions of unsaturated systems, including fluoroaromatic ones.26,27 Particularly in-plane distortions result in breaking of fragments’ equivalency in radical ions of symmetric molecules and in localization of spin density on one of the fragments.
The results of studying the reasons and regularities of appearing of structural distortions with symmetry decrease in radical ions of aromatic and unsaturated molecules are summarized in monographs.28,29 These data led to formation of new views on electronic and spatial structure of radical ions of polyfluoroaromatic molecules. Further development of ideas on structure of these ions is associated with non-empirical studies of adiabatic potential energy surfaces (PES), which led to the concept of structure flexibility.
Any deformation with symmetry decrease results in multi-well structure of the radical ion PES. In the simplest case of unique non-degenerate active mode Qu there are two equivalent PES minima (the result of distortion along В±Qu ), and connecting them saddle point corresponds to highly symmetrical structure of radical. When several active coordinates participate the number of stationary PES structures increases and the establishing of their interrelations becomes a nontrivial task, which requires a special study.
Manifestations of complex PES structure in chemical and physical properties of radical ions depend on the height of energy barriers and on the electronic structure of the system in minima. Therefore analyzing properties of the system revealing structure distortions with symmetry decrease is inseparably connected with studying of PES by modern quantum chemical methods.
III. Methodology of Potential Energy Surface Calculations
The following symmetry-based approach has been developed by us to study the PESes of fluoroarene radical ions.30,31 The primary geometry optimization is carried out within the symmetry of neutral precursor, that reflects the relaxation of formed radical ion along the totally symmetrical modes. At the geometry obtained single-point calculations of electron states belonging to different representations of the symmetry group are made and possible structure distortions resulting from their vibronic interaction are analyzed. Further geometry optimization of these states within the symmetry of neutral molecule leads to the location of stationary structures of radical ion which differ in symmetry of the ground electronic states. Additional single-point calculations with alteration of initial guess are carried out to identify whether the structure found lies on the ground state leaf. That allows us to reveal possible term crossings of electronic states belonging to different irreducible representations of the point symmetry group. The type of stationary PES points found is defined by the normal vibrations analysis. When imaginary vibrational frequencies are revealed we consider the symmetry reduction along the corresponding active mode. Method of internal reaction coordinate (IRC) is used for establishing of interrelations between the stationary PES points. The approach described allows us to establish the genesis and interrelations of stationary structures of radical ions and to build their total PES schemes.
The Hartree-Fock (HF)32,33 calculations lay down in the basis of our research being necessary for understanding the nature of the effects under study. We used the ROHF method implemented in the GAMESS program34 for the systems with open electron shell. Standard 6-31G basic set extended with polarization d-functions at heavy atoms (6-31G*) was utilized for radical cations. This basis set augmented by diffused sp-functions at the heavy atoms (6-31+G*) was used for anions. The electron correlation effects were taken into account at the CIS, RMP2, and CASSCF level of calculations. We used spin-unrestricted DFT method with the B3LYP functional for calculating the hfc constants and also for geometry optimization of stationary structures of radical ions.
The PES study for radical anions of benzene and its derivatives C6FnH6-n is complicated by the fact, that the molecules with n < 4 possess negative adiabatic electron affinity.6 When calculating anions of such system the inclusion of diffused functions into a basic set may lead to obtaining of the HF solutions corresponding to unbound states of free electron and neutral molecule.35,36 To select the bound (e.g. radical anion) solutions we applied the simplest stabilizing method – scaling the exponents of diffused basis functions.35 At that, it is proved, that ROHF/6-31+G* solutions of π-type on the contrary to σ-solutions are bound, the additional electron stays in valence region.
IV. Adiabatic Potential Energy Surfaces of the Benzene and Hexafluorobenzene Radical Ions
At first let’s consider radical ions of highly symmetrical (D6h) benzene and hexafluorobenzene molecules to trace the PES changes caused by fluorine atoms accumulation. Symmetrical D6h nuclear configuration, in which all four ions possess doubly degenerate ground state (2E1g: ...(1e1g)3(1e2u)0 for cations and 2E2u: ...(1e1g)4(1e2u)1 for anions), is unstable due to Jahn-Teller Effects (JTE). Active Jahn-Teller mode is degenerated as well and belongs to the e2g representation of the group. The form of adiabatic PES in this case (JTE E×e) is well known,37 it is the surface of conical intersection with three equivalent minima and three saddle points (Fig. 2). The stationary structures are interconnected by the pseudo-rotation coordinate. Moving along this coordinate allows the conical intersection point to be avoided. The section of adiabatic PES is showed in Fig. 3.
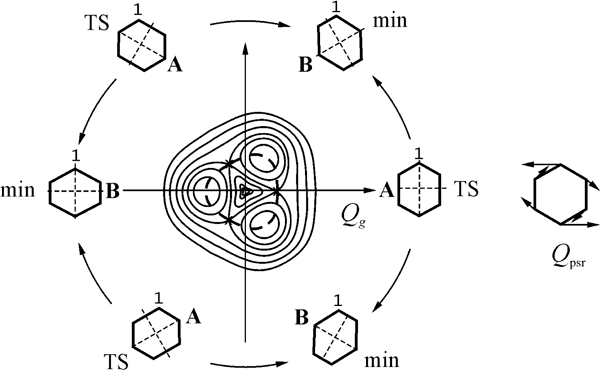
Figure 2. The view of adiabatic PES of the Jahn-Teller systems with C3 symmetry axis and pseudo-rotation scheme for planar D2h Structures of the benzene and hexafluorobenzene radical ions. Pseudo-rotation trough is marked with dotted line, to the right the corresponding coordinate is depicted.
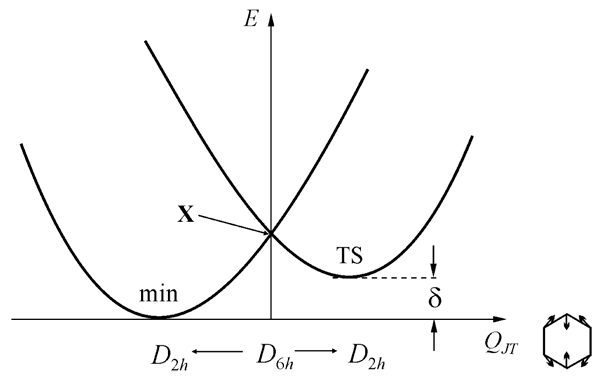
Figure 3. The section of PES from Fig. 2 along the totally symmetric in D2h group coordinate QJT (X – the point of conical intersection, δ – the height of pseudo-rotation barrier).
The distortion along the active coordinate leads to symmetry reduction to D2h and splitting the degenerate states. For every ion two D2h structures appear (distortions along ±QJT), compressed and elongated, which differ by the ground state symmetry. Later we will mark these structures as B and A, respectively (designations and SOMO view are showed in Fig.4). One of them corresponds to minimum, and another – to the transition state for pseudo-rotation.
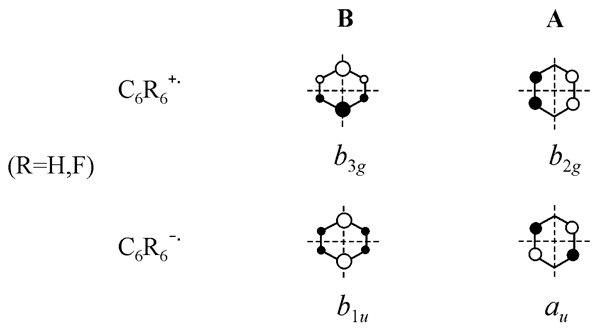
Figure 4. Designation for planar D2h structures of the benzene and hexafluorobenzene radical ions and a view of corresponding SOMOs.
In case of the benzene radical ions and hexafluorobenzene radical cation planar D2h structures correspond to the true minima and saddle points of PES. However radical anion of hexafluorobenzene as it has been mentioned above undergoes to out-of-plane deformations caused by the presence of low lying vibronically active excited Пѓ state (the Jahn-Teller pseudoeffect).
Successive symmetry reduction of the hexafluorobenzene radical anion along the active Jahn-Teller and pseudo-Jahn-Teller modes (e2g and e2u, respectively) has been considered by us on the basis of ROHF calculations taking into account the correlation corrections to energy at the RMP2 level.38 Combination of JTE and PJTE in C6F6— • results in doubling of a number of stationary PES points as compared to the JTE case alone (see Fig.2). Additional symmetry decrease along the out-of-plane active modes leads to structures of C2v and D2 symmetry. At that every planar structure produces two mirror-symmetrical non-planar ones. The interrelations of the non-planar structures have been established by the IRC calculations. The resulting scheme of pseudo-rotation in C6F6— • (Fig. 5) contain six minima (the structures of C2v symmetry) and six transition states (D2 structures), the height of barrier has been estimated as 0.2 kcal/mole at the RMP2/6 31+G*//ROHF/6 31+G* level of calculations. Energetically equivalent structures are interrelated by the S6 symmetry operation, and the diametrically opposite structures of pseudo-rotation cycle – by the inversion operation E* inverting spatial coordinates of all the particles.39 The way of pseudo-rotation is at the same time the way of inversion of the out-of-plane deformation.
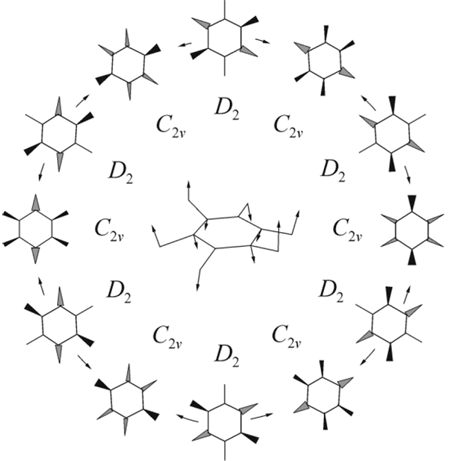
Figure 5. The scheme of pseudo-rotation in C6F6— •. In center the active coordinate is shown.
A principal particularity of the cycle obtained is the fact, that a smooth geometry variation along the pseudo-rotational coordinate is accompanied by continuous alteration of electron wave function, the phase jump typical for the pseudo-rotation schemes of the six planar structures of C6H6+/—. ions is absent.40 To elucidate the origin of that particularity let us set up a conformity of the every distorted structure of the pseudo-rotational scheme Fig. 5 with its generative planar D2h structure keeping in mind that the direction of out-of-plane fluorine shift is defined by the sign of electron transition density and supposing the phase factor to be constant (Fig. 6 a).
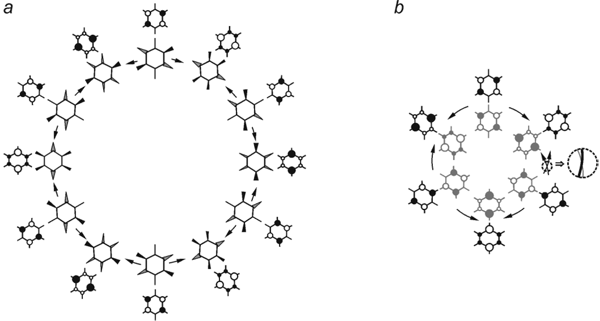
Fig. 6. Conformity of distorted and planar structures of C6F6— • (a) and schematic representation of the Möbius Band resulting from sewing of diametrically opposite planar structures of pseudo-rotation cycle which differ by the phase of wave function (b).
Pseudo-rotation cycle built in such way consists of 12 planar D2h structures and unlike the six structure cycle is characterized by the continuous change of electron wave function (Fig. 6 a). At that diametrically opposed planar structures (as well as produced by them non-planar ones) are interconnected by the operation of inversion, but due to the invariability of nuclear coordinates under action of the E* operation, they differ only in sign of wave function (SOMO phase), i.e. they are physically indistinguishable. According to a topological theorem,41 sewing all the diametrically opposed points of a circle of ring produces the Möbius Band. The result of such “sewing” (Fig. 6 b) is the known pseudo-rotation scheme of six planar structures of the benzene radical anion. Cutting the band along the medial line return us again to the twelve structure cycle. Taking into account the out-of-plane distortions we get exactly the C6F6— • case obtained early by the IRC procedure. Thus, pseudo-rotation schemes in C6H6— • and C6F6— • relate to each other as the Möbius Band and the result of its cutting along the medial line.
V. Potential Energy Surfaces of Radical Ions of Partially Fluorinated Benzenes
Pseudo-rotation in radical ions of benzene and hexafluorobenzene is the result of avoidance of ПЂ terms crossing in highly symmetrical D6h nuclear configuration (degenerated ground state). Asymmetrical substitution removes the degeneracy, but the fluorine atoms only slightly perturb the benzene ПЂ system. The energy splitting of degenerated in benzene ПЂ MO levels remains rather small in molecules of low-symmetrical (D2h, C2v) partially fluorinated benzenes.6 Therefore in their radical ions there are two close in energy ПЂ-states, in which unpaired electron occupied different components of degenerated in benzene MO. Respectively, for each ion there are two planar ПЂ structures,31,42 which differ by symmetry of the ground electronic state (orbital isomers), A (2A2) and B (2B1), in terms of C2v group. The difference of their energies О”EAB is minimal for radical ions of 1,2,3В trifluorobenzene, which ПЂ MO are quasi-degenerate and most sizeable for the ions of 1,4-F4C6H4 and 1,2,4,5-F4C6H2, with the maximal splitting of degenerate MO. Due to a weak perturbation of the benzene ПЂ-system by fluorine atoms, the SOMO of A and B structures of low-symmetric fluorobenzenes radical ions are similar to the ones displayed on the Fig.4.
Our analysis of the HF PES sections along the totally symmetrical coordinate connecting π structures has shown,31,42 that in all the cases besides 1,4-F2C6H4±• and 1,2,4,5-F4C6H2±•, the terms of π states are crossing. According to the data of calculations we can distinguish three main types of PES sections, which are displayed at Fig. 7 (the structure X corresponds to the point of π-terms crossing). The first case (ΔEAB < 3.5 kcal/mole), apart from the Jahn-Teller ions C6F6±• и 1,3,5-F3C6H3±, includes both ions of 1,2,3-F3C6H3. Radical ions 1,4-F2C6H4±• and 1,2,4,5-F4C6H2±•, which π terms do not cross, correspond to another limiting case Fig. 7 b (ΔEAB > 17 kcal/mole). The ions of all the other fluorobenzenes (5.5 < ΔEAB > 12 kcal/mole) belong to the intermediate case, Fig. 7 c. We shall note, that the type of PES crossing is defined by the relative position of fluorine atoms in the ring and doesn’t depend neither on their number, nor on the sign of the ion charge.
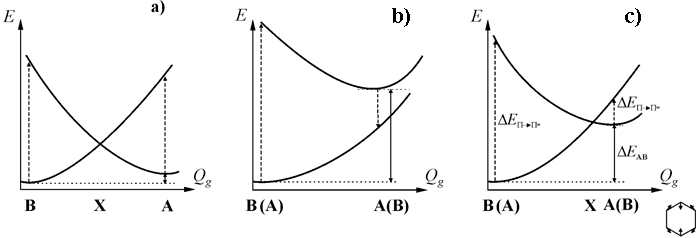
Fig. 7. Types of PES sections for the fluorobenzene radical ions along the Qg coordinate connecting A and B structures: limiting cases of small (a) and large (b) splitting of their levels (О”EAB) and intermediate case (c).
Thus, according to the HF calculations for the majority of fluorobenzenes their radical ions PESes are the surfaces of conical crossing. The crossing avoidance is carried out along the pseudo-rotation coordinate as in the case of Jahn-Teller ions. Symmetry of the coordinate (b2 in the C2v group) is defined by the symmetry of crossing terms. Out-of-plane deformations peculiar to radical anions make their PES structure more complicated that can be modeled by the corresponding cutting of the Möbius pseudo-rotation scheme similarly to the case of C6F6— •.
For radical ions of low symmetrical molecules the PES are less symmetrical and the barriers of pseudo-rotation are much higher (3-12 kcal/mole at the HF level of calculations), than for the Jahn-Teller systems. The accounting for electron correlation significantly influences both the height of the barriers and number of stationary structure of pseudo-rotation trough.31,42 We will consider the dependence of results on the calculation level by the example of radical ions 1,2,3-F3C6H3±• and 1,2,4,5-F4C6H2±•, which PES sections refer to the different types (Fig. 7 a and 7 b, respectively).
The fluorobenzene radical cations have a planar geometry, so their PES structure is fully determined by crossing of ПЂ states terms. In the case of radical cation of symmetric tetrafluorobenzene ПЂ terms do not cross both at the HF and post-HF calculation levels, so the surface is single-well.
For the 1,2,3-trifluorobenzene cation the terms cross, the crossing avoidance results in pseudorotation regardless of the calculations level.42,43 The accounting for electron correlation leads to decrease of a number of the stationary PES structures. With the correction for zero point vibration energy the PES consists of one minimum and one transition state (Table 1). The barrier for pseudo-rotation is quite low, about ~1.5-2 kcal/mole (Table 1), that points out the structural flexibility of the radical cation and its possible spectral manifestation. Unfortunately, this radical cation is out of consideration by the ESR method.44 However, the unusual structure of this cation had been mentioned by Miller, Bondybey and colleagues45,46 during studying of the laser excitation and fluorescence spectra.
Table 1. Dependence of the 1,2,3 F3C6H3+• PES shape on the calculation level (the total energies are given in a.u. and relative ones are in kcal/mole).
|
Method |
Structure |
Etot |
Na |
Erel |
ErelZPE |
|
ROHF/6-31G* |
C2v (2B1) |
-526.928356 |
0 |
0 |
0 |
| |
C2v (2A2) |
-526.923255 |
1 |
3.20 |
1.05 |
| |
Cs (2A") |
-526.925137 |
0 |
2.02 |
1.63 |
| |
Cs (2A") |
-526.924458 |
1 |
2.45 |
0.75 |
|
CASSCF(5,5)/6-31G* |
C2v (2B1) |
-526.975333 |
1 |
0.15 |
0 |
| |
C2v (2A2) |
-526.972963 |
1 |
1.64 |
1.40 |
| |
Cs (2A") |
-526.975570 |
0 |
0 |
0.74 |
|
RB3LYP/6-31G* |
C2v (2B1) |
-529.356997 |
0 |
0 |
0 |
| |
C2v (2A2) |
-529.353080 |
1 |
2.46 |
1.28 |
a Number of imaginary vibrational frequencies
Four types of non-planar stationary structures (Fig. 8) – two minima (structures Cs and C1(B)) and two saddle points (C2 and C1(A) structures) [For the structures of C1 symmetry the SOMO type is mentioned within brackets (A ~ a2, B ~ b1)], which correspond to transition states of pseudo-rotation 31,47.48 exist at the 1,2,3 F3C6H3— • PES regardless of calculations level. Pseudo-rotation scheme in this radical anion combines twelve structures. It is analogue to the one displayed at Fig. 5 for C6F6— •, but unlike that hydrogen atoms remain in the plane of the benzene ring and minima as well as saddle points are non-equivalent in energy. Pseudo-rotation barrier is rather low (Table 2). Thus, both ions of 1,2,3-trifluorobenzene are structure flexible.
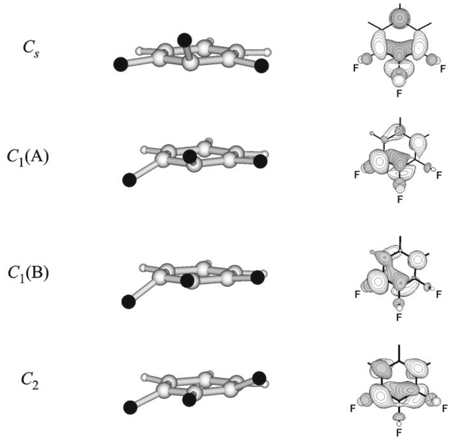
Fig. 8. Stationary PES structures of the 1,2,3-trifluorobenzene radical anion and their SOMO images.
In case of 1,2,4,5-F4C6H2— • the π-states terms of planar structures do not intersect, but both planar structures are unstable towards out-of-plane distortions. The PES again is a pseudo-rotation surface resulting from crossing of the potential surfaces of different symmetry non-planar structures. Pseudo-rotation cycle (Fig. 9) is formed by two equivalent minima (the structures of C2v symmetry) and two transition states (D2 structures). The peculiarity of this anion is significant out-of-plane shifts of the H atoms in the minimum energy structure.
Table 2. Stationary structures of 1,2,3-trifluorobenzene radical anion (the total energies are given in a.u. and relative ones are in kcal/mole).
|
Method |
Structure |
Etot |
N a |
Erel, |
ErelZPE |
|
ROHF/6-31+G* |
Cs |
-527.204096 |
0 |
0.93 |
0.92 |
| |
C1(A) |
-527.198222 |
1 |
4.62 |
3.62 |
| |
C1(B) |
-527.205581 |
0 |
0 |
0 |
| |
C2 |
-527.195525 |
1 |
6.31 |
5.19 |
|
RMP2/6-31+G* |
Cs |
-528.556354 |
|
0 |
- |
| |
C1(A) |
-528.550305 |
|
3.80 |
- |
| |
C1(B) |
-528.550590 |
|
3.62 |
- |
| |
C2 |
-528.549831 |
|
4.09 |
- |
|
UB3LYP/6-31+G* |
Cs |
-529.709613 |
0 |
0 |
0 |
| |
C1(A) |
-529.705929 |
1 |
2.31 |
1.81 |
| |
C1(B) |
-529.706811 |
0 |
1.76 |
1.68 |
| |
C2 |
-529.703361 |
1 |
3.92 |
3.13 |
a Number of imaginary vibrational frequencies
The mirror-symmetrical C2v structures are related not only by pseudo-rotation, but by direct inversion (via transition states C2h symmetry) as well, the second way is more favorable in energy. As opposed to pseudo-rotation barrier the barrier for direct inversion increases at going to post-HF methods, remaining at the same time lower than the pseudo-rotation barrier (Table 3).
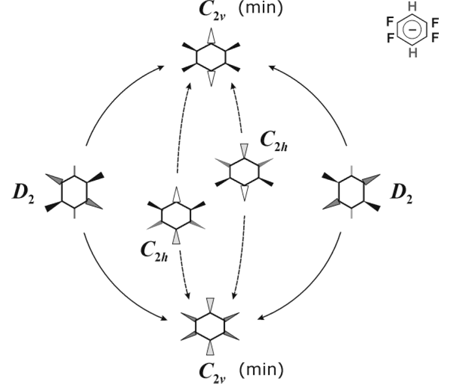
Fig. 9. The scheme of potential energy surface for the 1,2,4,5-F4-C6H2— • Radical Anion.
Table 3. Stationary structures of the 1,2,4,5-tetrafluorobenzene radical anion (the total energies are given in a.u. and relative ones are in kcal/mole).
|
Method |
Structure |
Etot |
N a |
Erel, |
ErelZPE |
|
ROHF/6-31+G* |
C2v(z) |
-626.064651 |
0 |
0 |
0 |
| |
D2 |
-626.044396 |
2 |
12.71 |
11.72 |
| |
C2(y) |
-626.047091 |
1 |
11.01 |
10.35 |
| |
C2h |
-626.063785 |
1 |
0.54 |
-0.37 |
|
UB3LYP/6-31+G* |
C2v(z) |
-628.919899 |
0 |
0 |
0 |
| |
D2 |
-628.911831 |
2 |
5.06 |
4.26 |
| |
C2(y) |
-628.919391 |
1 |
4.71 |
4.31 |
| |
C2h |
-628.915403 |
1 |
2.82 |
2.28 |
|
RMP2/6-31+G* |
C2v(z) |
-627.544517 |
0 |
0 |
- |
| |
D2 |
-627.537945 |
1 |
4.12 |
- |
| |
C2h |
-627.541180 |
1 |
2.09 |
- |
a Number of imaginary vibrational frequencies
The limiting situations considered point out that pseudo-rotation in radical ions of low-symmetrical partially fluorinated benzenes is kept at the post-HF calculation level as well. All anions of this row and some cations are structure flexible towards coordinate of pseudo-rotation.
The accomplished researches revealed the reason for similarity and differences in the PES structures for radical cations and anions of fluorinated benzenes.49,50 Their common features are connected with weak disturbance of the degenerate frontier benzene π MO by the fluorine atoms that leads to proximity of the resulting π state levels and opportunity for their terms crossing. The main reason for the differences is a strong perturbation of the benzene σ system by fluorine atoms, which results in lowering the levels of excited σ-states in anions and their rising in cations. This leads to instability of planar nuclear configurations of radical anions, while cations keep their planar structure. In radical anions of fluorinated benzenes the out-of-plane deformations of orbital isomers A and B produce structures belonging to different point groups of symmetry. In case of C6H5F— • used as a model, pseudo-rotation was proved to be the result of avoided crossing of potential surfaces different by symmetry of non-planar configurations along a three-dimensional coordinate.49 Therefore it exists regardless of π terms crossing for planar structures. Thus, all the fluorobenzene radical anions are structure flexible in a varying degree.
VI. Manifestation of Structure Flexibility in the ESR Spectra of Polyfluoroarene Radical Ions
According to the results of quantum chemical PES researches described above the fluorinated benzenes’ radical anions are structurally flexible towards the coordinate of pseudo-rotation. The significant redistribution of spine density in the process of pseudo-rotation, in which structures of different spatial symmetry participate (See Fig. 8) allows expecting the fact, that predicted by calculations structure flexibility will display itself in the form of temperature dependence of hyperfine interactions in ESR spectra. Thus, experimental checking the results obtained is possible.
The necessary condition for that is a reliable description of hfc constants with 19F nuclei in non-planar radical anions of pseudo-π-type. According to the results obtained for C6F5H— • and C5NF5— • anions,50 which relatively high barriers for pseudo-rotation (~5 and 11 kcal/mole, respectively) allow considering, that spectra do not virtually depend on temperature, and experimental values aF are well reproduced by the UB3LYP/6-31+G* calculations (Table 4). Taking into consideration the sensibility of aF values towards structural parameters we can also speak about adequate description of the radical anion geometries at that level of calculations.
Table 4. Calculated (UB3LYP/6-31+G*) for structures of minimal energy (Cs) C6F5H— • and C5NF5— • and experimental51-53 constants aF (G).
|
Radical Anion |
Nucleus |
Calculation |
Experiment |
|
|
matrix |
solution |
|||
|
C6F5H— • |
19F (o-) |
54.2 |
45.5 |
48.7 |
| |
19F (m-) |
112.6 |
101.0 |
107.0 |
| |
19F (p-) |
268.8 |
279.0 |
295.0 |
|
C5NF5— • |
19F (О±-) |
19.5 |
17.4 |
|
| |
19F (ОІ-) |
69.0 |
61.2 |
|
| |
19F (Оі-) |
255.5 |
249.0 |
|
| |
14N |
3.4 |
3.6 |
|
We choose non-described earlier in literature radical anion of 1,2,3-trifluorbenzene, for which the height of pseudo-rotation barrier is minimal in the row of fluorinated benzenes anions as an object of experimental research by the optical detected (OD) ESR method. Experiments specially conducted by V’ushkova, Bagriansky and Molin proved, that 1,2,3 F3C6H3— • spectrum depended on temperature.47,48 At increasing of temperature a significant shift of spectrum lines occurs without their widening, that is typical for fast spectral exchange.54
We have used the results of IRC calculations with UB3LYP/6-31+G* method to interpret the observed changes in spectrum (Fig.10). The averaging of calculated along the pseudo-rotation trough values of 19F hfc constants within the range of classic Boltzman distribution reproduces well temperature dependence of ESR spectrum observed within the temperature interval of 243-325 K (Fig. 11). Thus, experimental confirmation of 1,2,3 F3C6H3— • anion structure flexibility towards the coordinate of pseudo-rotation had been obtained.
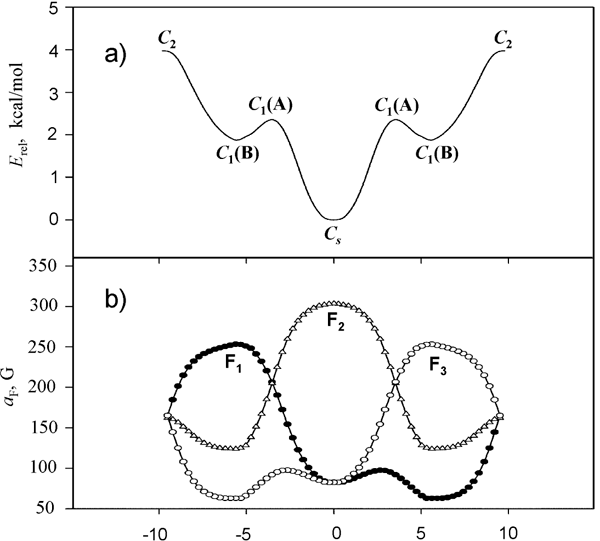
Fig. 10. Changes in energy (a) and in 19F hfc constants (b) of the 1,2,3- F3C6H3— • radical anion during moving along pseudo-rotation coordinate from the C2 transition states to Cs minima (UB3LYP/6-31+G*).
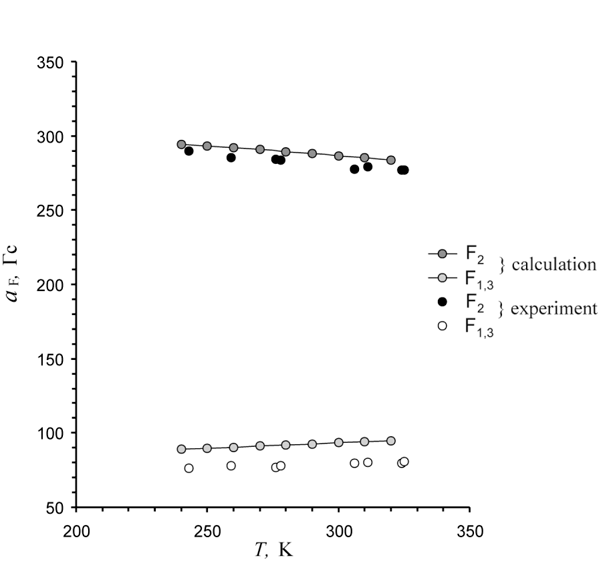
Fig. 11. Temperature dependence of 19F hfc constants in the 1,2,3 F3C6H3— • radical anion by the data of OD ESR experiments and calculations in classic model.
Latter it has been proved, that structure flexibility was not an exclusive feature of radical anions of benzene row. The perfluorobiphenhyl radical anion (C6F5)2— • recently registered by OD ESR method exemplifies the structure flexible system, which ESR spectrum is not temperature dependent.55 Experimental values of aF constants, 72 (2F), 26 (4F) and 18.6 (4F) G, definitely point out the deviation of fluorine atoms from rings’ plane. Taking into account that LUMO of the perfluorobiphenyl molecule is a symmetrical combination of b1 rings’ orbitals it could have been expected, that during capture of electron the out-of-plane deviation is mostly significant for F para-atoms, the corresponding symmetry reduction is D2 → C2. However, the model of non-planar C2 structures don’t explain the observed aF values.
The PES study carried out for interpretation of the OD ESR data proved55,56, that (C6F5)2— • combines all three types of structural distortions considered: avoidance of conical crossing, out-of-plane deviations of fluorine atoms, and disturbance of the phenyl rings equivalence. The reason for the last type of distortions is significant non-coplanarity of the rings due to steric interaction of ortho-fluorine atoms.
The energy minimum corresponds to the totally asymmetric C1 structure (Fig. 12) where aromatic ring planes are twisted at ~45Лљ, the C-F bonds out-of-plane deviations in one of the rings being more than in the other. The unpaired electron density is located in the more distoreted ring.
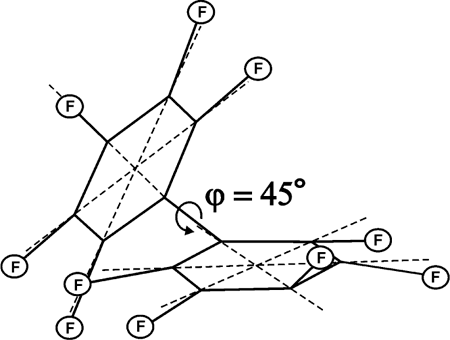
Fig. 12. Minimum energy structure (C1) of the perfluorobiphenyl radical anion.
The radical anion PES is formed by two cycles (Fig.13) composed of four equivalent minima (C1 structures) divided by transition states of C2 symmetry (structures of different cycles differ by the П† angle sign), the height of barriers is ~0.5В kcal/mol. These cycles are connected by higher in energy (~4.5 kcal/mol) transition states of Cs symmetry.
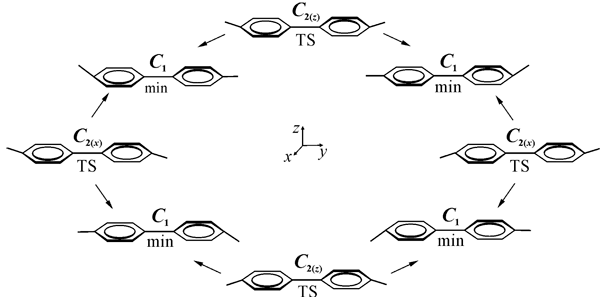
Fig. 13. Interrelation of the (C6F5)2— • low-energy structures.
Extremely small height of barriers within each cycle leads to total hfc averaging under conditions of OD ESR experiments (liquid solution). Averaging of calculated by UB3LYP method aF constants over the C1 structures leads to values 71.3В (2FВ para-), 18.9В (4FВ orthoВ ), and 28.1В (4FВ meta-) G, which agree well with experimental ones, 72В (2F), 18.6В (4F) and 26В (4F) G.
Thus the results of systematic PES studies for polyfluorarene anions row were confirmed experimentally that proved the adequacy of the models used. This points out that the complex PES structure must be considered when interpreting the data of spectral and chemical experiments.
VII. Structural Distortions and Monomolecular Cleavage of Haloarene Radical Anions
Our researches proved that structure distortions with symmetry lowering are typical for radical ions of polyfluoroaromatic molecules. Such distortions result in qualitative change of electron density distribution which able to influence the passing of chemical reactions. Energy barriers dividing the PES minima are in essence the barriers for electron density intramolecular transfer. Thus, pseudo-rotation in the benzene row radical ions is a mechanism of unpaired electron density transfer between the ring positions. So we can expect, that positional selectivity of reactions of these radical ions will be to a great extent defined by their structure flexibility towards the pseudo-rotation coordinate. Out-of-plane distortions typical for radical anions of polyfluoroaromatic compounds are the mechanism of electron density transfer between ПЂ and Пѓ systems. Structural deformations of this type may be of a principal importance for the reactions forbidden by symmetry.
We have studied57-61 the connection between the PES structure and reactivity using the example of typical reaction of haloaromatic radical anions – fragmentation with the halide ion elimination – a key stage of synthetically important reactions of nucleophilic substitution (SRN1) and reductive dehalogenation. For planar π radicals the reaction is forbidden by symmetry,62 its barrier is connected with crossing of π and σ states terms:
ArHal — • (π radical) → Ar• (σ radical) + Hal—.
It had been supposed,62 that transfer from ПЂ onto Пѓ term was realized owing to vibrations.
The fluoride ion elimination occurs only in polar solvents, the direct calculations of the reaction paths require the solvation to be taking into account. The alternative approach – modeling of transition state by the gas-phase calculations – was applied to studying the decomposition of fluorinated benzonitriles and benzoates radical anions.57,58
The offered model of transition state is based on idea of avoiding the ПЂ and Пѓ terms crossing due to out-of-plane distortion accompanying the C-F bond stretching. In this model energetic barrier for F Л‰ elimination, Ea, is determined by the position of ПЂ-Пѓ-crossing point, rCFx, stabilization energy at this point owing to out-of-plane distortion, the transition state energy lowering due to in-plane relaxation of the ring, and the difference of solvation energies of transition state and initial anion (the last contribution is supposed to be constant in the row of related radicals).
Fulfilled in this model semi-empirical INDO calculations allowed explaining the experimentally observed regularities of cleavage of the fluorinated benzonitriles and benzoates radical anions, including dramatic (about six orders of magnitude) increase of the reaction rate constant with fluorine atoms accumulation. In both cases calculations led to satisfactory linear dependence between barriers of Ea reaction, estimated with the model transition states and experimental values of log kd (the decay rates constant). The results for full row of fluorobenzoate radical anions are shown at Fig. 14, specific solvation in aqueous solutions has been modeled by calculations of corresponding benzoic acids).
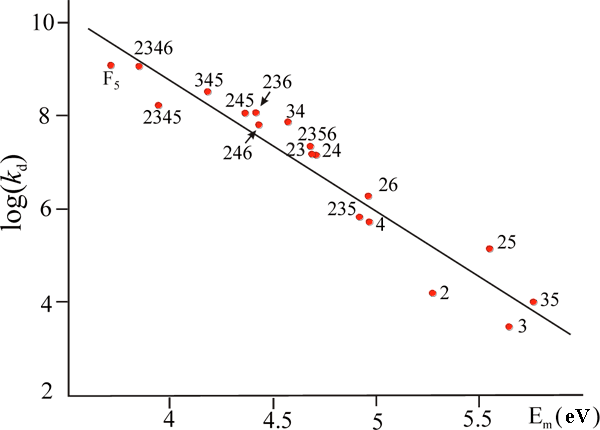
Fig. 14. Relationship between the model reaction energy barrier Ea for the F‾ elimination from the fluorobenzoic acids radical anions (UHF/INDO) and the experimental fragmentation rate constant kd of radical anion of corresponding benzoates; R = 0.96 (fluorine atom positions are marked with digits).
Within the same ideology the value of energy splitting between the ground ПЂ and excited Пѓ- state levels for the radical anion planar structure (О”EПЂПѓ) responsible for out-of-plane deformation was offered to be used as an index of reactivity.57,В 58 It had been proved, that О”EПЂПѓ values may be applied for both qualitative analysis of relative ability of anions to eliminate fluoride-ion and explaining the reaction regioselectivity.
Using this index approach we managed to interpret the data on regioselectivity of the polyfluoroxylenes hydrodefluorination under the action of metals (Fig. 15) supposing the intermediate radical anions formation and their further fragmentation.
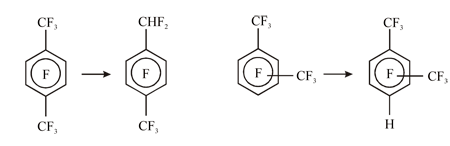
Fig. 15. Scheme of main transformations of isomeric perfluoroxylenes under the action of Zn(Cu)-DMF-H2O.
According to the results of INDO calculations,59 the energies of ПЂв†’Пѓ* excitation for radical anions of perfluoro-ortho- and metha-xylenes are lower as compared with the para-isomer. Besides that, the positions of preferred localization of ПЂ and Пѓ* SOMO density for perfluoro-para-xylene anion do not coincide (e.g. the electron transition density is small). So, for this radical anion dissociation is more likely for the C-F bond in benzyl position. On the contrary, in case of perfluoro-metha- and ortho-xylols anions SOMO of the both ПЂ, and Пѓ* states are localized preferably at C-F bonds of aromatic ring, as a result the dissociation of these bonds occurs keeping C-F bonds in benzyl position.
The results of semi-empirical studies57,58 point out the essentially non-planar structure of transition states for the fluoroarene radical anions cleavage. Similar structure distortions related with the π-σ crossing avoidance can be expected also for π radical anions of other haloaromatic molecules as well. It is impossible to localize the true transition state of fragmentation for fluorine containing anions without direct including of solvent, as in the gas phase σ-term is not dissociative. At the same time the chloride-ion elimination is observed in gaseous phase as well.63 C6H5Cl— •anion is a convenient model for theoretical studying of C-Hal bond heterolytic dissociation in haloaromatic radical anions.64
According to the results of ROHF/6-31+G* study of the C6H5Cl — • potential energy surface the reaction mechanism depends on what anion state results from electron capture. Thus, in 2A1 σ-state the chloride-ion elimination is symmetry allowed and is carried out without any barrier. At electron capture at 2B1 π-term – the Cl ˉ elimination from the position of maximum unpaired electron density – reaction coordinate includes out-of-plane deviation of C-Cl bond (π-σ crossing avoidance). In case of C6H5Cl— • as a result of strong vibronic interaction the crossing avoiding is carried out with no barrier. At the most likely electron capture onto 2A2 term the decomposition has got a double forbidding by symmetry. The additional forbidding is connected with impossibility of transfer onto repulsive σ–term from the state with zero density of unpaired electron at C-Cl bond. The reaction coordinate includes in-plane deformation of the ring (avoiding of π-terms crossing along the pseudo-rotation coordinate), at that the add electron density is transferred onto C atom connected with Cl.
The transition state of chloride-ion elimination found for the first time by calculations has got non-planar structure and possesses no symmetry elements peculiar to the initial C6H5Cl (C2v) molecule.60 The symmetry elements absence is caused by combination of in-plane deformation of the ring and out-of-plane deviation of the breaking bond. The reaction barrier is connected with the π terms crossing avoidance along the pseudo-rotation coordinate. A negligibly small barrier height (0.25 kcal/mole at MP2/6 31+G*//ROHF/6 31+G* calculations level) comes from the C6H5Cl— • structure flexibility towards pseudo-rotation coordinate and conforms to the idea on synchronous stages of electron transfer and C-Cl bond cleavage in C6H5Cl — •.64
Described ways of the C6H5Cl — • model anion fragmentation60 reflect possible mechanisms of dissociative cleavage of haloaromatic radical anions. As it has been mentioned before, it is impossible to locate the true transition state for fluoride-ion elimination without direct including of solvent in calculations. However, from considerations of symmetry, it can be stated, that the decay of C6H5F — • (the ground electronic state is 2A2) in polar media can be carried out only within the mechanism revealed that includes electron density transfer owing to pseudo-rotation and out-of-plane deviation of breaking bond. It can be supposed, that in case of C6H5F — • as well the main factor determining passing of the reaction is electron transfer to the breaking C-F bond. This prediction was confirmed by Pieriny and Vera65 during studying of energy profiles of C-Hal bonds dissociation with the UB3LYP method taking into account the solvent effects within the CPCM model: in case of C6H5F — • the transition to σ*-term occurs not from the ground state, but from the excited π*-state, which SOMO have no nodal plane passing through the C-F bond. According to Pieriny and Vera, 65 the radical anion decomposition occurs from 2B1-state virtually barrier-less. Thus, the barrier for monomolecular fragmentation is the barrier for transition from one π- term to another that is the pseudo-rotation barrier in our terminology.
The dependence of mechanism of the C-Hal bond heterolytic breakdown in ПЂ type radical anions on the density of add electron on the breaking bond has been studied by us at the ROHF, RMP2//ROHF, and B3LYP levels of calculation by the example of radical anions of isomeric chlorobenzonitriles Cl-C6H4CN.61 The presence of acceptor CN-group stabilizes the anions in regards to chloride-ion elimination, and also fixes the b1-type SOMO with preferable localization para->В orthoВ В >В metha regardless of the position of Cl atom which weakly perturbs ПЂ system (Fig. 16).
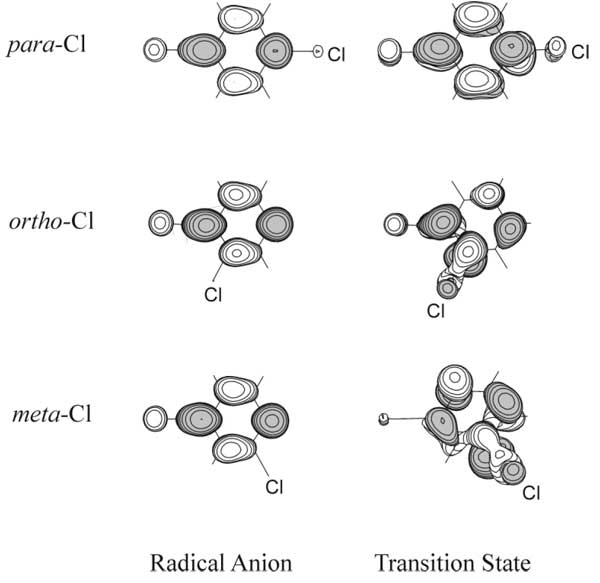
Fig. 16. SOMO images for initial Cl-C6H4CN — • radical anions and for transition states of their fragmentation with Cl—elimination.
For all the three isomeric anions the transition states of fragmentation are characterized by significant out-of-plane deviations of C-Cl bond. Maximum deviation is observed for meta-, and minimum – for para-isomer. For the para-isomer the situation corresponds to the fragmentation of C6H5Cl — • model anion at electron capture onto 2B1 term: reaction coordinate is a superposition of C-Hal bond stretching and its out-of-plane deviation. Transition state has got a Cs(yz) symmetry.
In the initial radical anion structures of ortho- and meta-isomers the SOMO density is also located in para- and ipso-positions relative to CN group, while in the transition states of fragmentation it is significantly shifted to C-Cl bond (Fig. 16). Moving along the pseudo-rotation coordinate serves as the mechanism of electron density transfer between the ring positions. The sequence of calculated reaction barrier heights (para- > ortho- > meta-) doesn’t depend on the level of calculation and corresponds to sequence of experimental rate constants of radical anions cleavage in liquid ammonia:66 The change of relative ability of para- and ortho- isomers to eliminate chloride-ion which is observed in going to more polar solvent (H2O+tBuOH)67 may be explained by the different extent of a negative charge localization at the leaving chlorine atom in the fragmentation transition states found.
The considered effects of terms crossing avoidance and intramolecular electron density transfer owing to structure distortions seem to play a significant role in mechanisms of others symmetry forbidden reactions. The structure distortions arising in course of such reactions are determined by symmetry and so exist in both gaseous phase and solvent and don’t depend on calculation method. Note that analysis of term crossings possible in a reaction course is rather useful for predictions of the reaction transition state structure.
References
Recommended for publication by Prof. V.E. Platonov
Fluorine Notes, 2013, 87, 1-2
