Received: December, 2012
Fluorine Notes, 2013, 87, 7-8
The Synthesis of Pentafluoroacetonyl Derivatives based on Iodopentafluoroacetone
N.D. Kagramanova, A.A. Tyutyunovab, S.R. Sterlina
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, V-334, GSP-1, 119991 Moscow, Russia
b"P&M-Invest", ul. Vavilova 28, 119991 Moscow, Russia
e-mail: tuytuynov@rambler.ru
Abstract: Iodopentafluoroacetone smoothly undergoes dimerization in the presence of Cu-powder yielding perfluoroacetonylpropenyl ether. The adducts of iodopentafluoroacetone with ethylene and tetrafluoroethylene were obtained under the conditions of thermal initiation. The transformations of 2-(perfluoroacetonyl)ethyl iodide have been studied.
Keywords: iodopentafluoroacetone, ethylene, tetrafluoroethylene.
Earlier it was found that the thermolysis of iodopentafluoroacetone (1) (autoclave, 250°C/25 h) led to the formation of perfluoroacetonylpropenyl ether (2) – the product of O-C-dimerization of 1 [1]. The authors assumed that the reaction proceeded according to the scheme comprising the homolytic dissociation of C-I-bond in 1 with subsequent addition of perfluoroacetonyl radical (3) to oxygen atom of carbonyl group of another molecule 1 accompanied by elimination of iodine radical from ICF2-group.
In spite of the expected easiness of the homolysis C-I-bond in 1, that should lead to the generation of relatively stable heteroallyl radical 3, the reaction conditions appeared to be very severe that was most probably connected with the reversible character of homolytic rupture.
In the given work we have found that in the presence of Cu-powder as iodine acceptor acetone 1 formed dimer 2 in quantitative yield at 90-100В°C.
Scheme 1
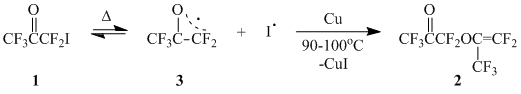
The easiness of acetone 1 homolysis allows to apply the radical addition reactions of 1 to unsaturated compounds as a convenient method of introducing perfluoroacetonyl moiety into different classes of organic compounds.
Indeed, under the conditions of thermal initiation acetone 1 adds smoothly to ethylene affording adduct 4 – a starting material for preparing oxolane 5 by intramolecular cyclization of iodide 4 in the presence of CsF. The saponification of 4 under the conditions described in [2] resulted in formation of oxyoxolane 6 – the product of intramolecular cyclization of initially formed alcohol 6a.
Scheme 2

The dehydration of oxyoxolane 6 under the action of P2O5 led to olefine 7. Evidently alcohol 6a is the immediate precursor of ketoolefine 7, and in order to shift the equilibrium 6 ↔ 6a to the right the reaction mixture was heated up to ~300°C by Bunsen burner.
Scheme 3

The reaction of 1 with tetrafluoroethylene (autoclave, 205В°C) gave adduct 8 (Scheme 4), but in this case 1,2-diiodotetrafluoroethane (9) was formed as a by-product. We failed to isolate analytical sample of adduct 8 from azeotrope 8-9. The structure of compound 8 was established on the basis of CMS and 19F NMR data.
Scheme 4
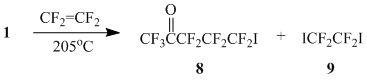
Thus the above examples show that the radical addition of iodopentafluoroacetone to nonfluorinated and fluorinated ethylenes and further transformations of the adducts obtained poses convenient method of introducing perfluoroacetonyl group in to different classes of organic compounds.
Experimental
1H, 19F NMR spectra were recorded on Bruker AVANCE-300 spectrometer at 300 and 282 MHz, accordingly; the external standard was CDCl3 or D2O. Chemical shifts for 1H spectra are presented vs. the residual signal of the solvent (Оґ 7.25; 4.79) and are given in ppm vs. tetramethylsilane. Chemical shifts in 19F spectra are given in ppm vs. CF3CO2H. Downfield shifts are positive. The Raman spectra were recorded on Jobin Yvon LabRam Raman spectrometer. The mass spectra were recorded on Finnigan Polaris Q (Trace GC ultra) Mass spectrometer.
Iodopentafluoroacetone (1) was obtained by method [3].
Dimerization of iodopentafluoroacetone in the presence of Cu-powder
The mixture of iodopentafluoroacetone (1) (30 g, 0.109 mol) and Cu-powder (7 g, 0.11 g-atom) was heated at 100В°C/3 h in a sealed glass tube. The reaction product 2 (16.1g, quantitative yield) was evacuated under reduced pressure into receiver (-78В°C). 19F NMR-spectrum of the product obtained corresponded of that described in [1]. Further distillation gave 15.5 g of 2, b.p. 69-70В°C (lit. data: b.p. 65-72В°C [1]).
The reaction of iodopentafluoroacetone with ethylene
The mixture of acetone 1 (25 g, 91 mmol) and ethylene (6 l, 240 mmol) was shaken in 100 ml stainless steel autoclave at 200В°C/10 h, then the liquid part of reaction mixture (27 g) was distilled to give 24.6 g (89%) of adduct 4, b.p. 70-79В°C (85 Torr).
The analytical sample was isolated by redistillation, b.p. 71-72В°C (85 Torr). Found (%): C, 19.86; H, 1.33; F, 31.46. C5H4F5IO. Calculated (%): C, 19.85; H, 1.33; F, 31.23. IR-spectrum (ОЅ, СЃm-1): 1788 (C=O). 1H NMR Оґ: 2.53, (s, 2H, CH2I), 2.98, (s, 2H, CF2CH2); 19F NMR Оґ: -1.83, (s, 3F, CF3), 31.25 (s, 2F, CF2).
The preparation of oxolane 5
The mixture of ketoiodide 4 (9 g, 30 mmol), CsF (5 g, 33 mmol) and 25 ml of diglyme was stirred at 35-40В°C/6 h, filtered, the filtrate was washed with dilute aq. HCl, extracted with ether, extract was dried MgSO4, the ether was evaporated to give a residue (3.9 g), which contained compounds 4 : 5 = 1 : 9. Its distillation afforded 3.5 g of a fraction (106-135В°C) that contained ~95% of oxolane 5 (GLC, 19F NMR), yield 75% based on 4 entered the reaction.
The analytical sample of 5 was obtained by redistillation, b.p. 114-115В°C. Found (%): C, 30.72; H, 2.06; F, 58.43. C5H4F6O. Calculated (%): C, 30.92; H, 2.06; F, 58.76. 19F NMR Оґ: 2.45 (dt, 3F, 3JCF3-CF = 13 Hz, 4JCF3-CF2 = 2.9 Hz, CF3), AB-pattern with centrum 36.35 (2F, JAB = 245.3 Hz, CF2), 44.8 (br.s, 1F, CF).
The synthesis of oxyoxolane 6
The mixture of ketoiodide 4 (24.6 g, 81.46 mmol), 3.8 g (210 mmol) H2O and 160 g NMP was stirred at 150В°C/13 h, then the reaction mixture was poured into 400 ml of water, extracted with ether (4x50 ml), extract was evaporated, the residue was distilled under reduced pressure (60-110В°C (17 Torr)) to give 19 g of a fraction, which was redistilled yielding 12.5 g (78%) 6, b.p. 72-98В°C (30 Torr).
Found (%): C, 30.27; H, 2.77; F, 48.12. C5H5F5O2.0.25H2O. Calculated (%): C, 30.53; H, 2.80; F, 48.35. 1H NMR Оґ: 2.25Г·2.40 (m, 2H), 3.94Г·4.01 (m, 2H). 19F NMR Оґ: 3.93 + 3.94 (d, 3F, JCF3-OH = 14 Hz, CF3), AB-pattern with centrum 36.5 (2F, JAB = 240.6 Hz, CF2). Mass-spectrum 6 (m/z, reference): 193 [M+H]+; 175 [M-OH]+; 153 [C5H4F3O2]+; 148 [C3HF5O]+; 135 [C5H2F3O]+; 123 [M-CF3]+; 103 [C4H4FO2]+; 95 [C3H5F2O]+; 83 [C4H3O2]+; 77 [C3H3F2]+ (100%); 75 [C3HF2]+; 69 [CF3]+; 64 [C2H2F2]+; 55 [C3F]+; 51 [CHF2]+; 45 [C2H5O]+; 39 [C3H3]+.
The preparation of ketoolefine 7
The mixture of oxyoxalane 6 (13.2 g, 70 mmol) and P2O5 (9.5 g, 77 mmol) was heated by Bunsen burner, collecting the distillate into receiver cooled with ice. The distillation of distillate afforded 6.1 g (51%) of ketoolefine 7, b.p. 48-50В°C.
Found (%): C, 34.65; H, 1.67; F, 54.88. C5H3F5O. Calculated (%): C, 34.48; H, 1.72; F, 54.60. IR-spectrum (ОЅ, cm-1): 1651.0 (C=C); 1788.6 (C=O). 1H NMR Оґ: 6.27Г·6.55 (m). 19F NMR Оґ: 1.96 (s, 3F, CF3), 30.38 (s, 2F, CF2).
The reaction of iodopentafluoroacetone with tetrafluoroethylene
The mixture of acetone 1 (29 g, 106 mmol) and tetrafluoroethylene (3.25 l, 130 mmol) was shaken in 100 ml stainless steel autoclave at 205°C/11 h. The liquid part of the reaction mixture (34.1 g) contained 10% of starting ketone 1, 20% of 1,2-diiodotetrafluoroethane (9) and 70% of γ-hexafluoropropyltrifluoromethyl ketone (8) (yield 68% based on 1 entered the reaction). The further distillation gave 30.5 g of a fraction (98-115°C), which contained compounds 8 : 9 = 78 : 22 couldn’t be divided by rectification. The structure of adduct 8 was established according to CMS and 19F NMR data.
19F NMR Оґ: -17.8 (s, 2F, CF2I), 1.5 (s, 3F, CF3), 36.5 (s, 2F, C(O)CF2), 40.5 (s, 2F, CF2). Mass-spectrum 8 (m/z, reference): 375 [M+H]+; 355 [M-F]+; 277 [C3F6I]+; 247 [M-I]+ (100%); 239 [C3F4I]+; 227 [C2F4I]+; 208 [C2F3I]+; 181 [C4F7]+; 177 [CF2I]+; 169 [C3F7]+; 131 [C3F5]+; 100 [C2F4]+; 97 [C2F3O]+; 69 [CF3]+.
References
- A.Yu. Volkonskii, G.G. Bargamov, E.M. Rokhlin, Russ. Chem. Bull., 1989, 38 (9), 1972-1974.
- T. Hayashi, M. Matsuo, US Pat. № 4001309, 1977.
- S.A. Postovoi, I.M. Vol'pin, E.I. Mysov, Yu.V. Zeifman, L.S. German, Russ. Chem. Bull., 1989, 38 (5), 1067-1070.
Recommended for publication by Prof. S. Sterlin
Fluorine Notes, 2013, 87, 7-8
