Received: ноябрь 2012
Fluorine Notes, 2013, 86, 7-8
Структурные особенности, Теория функционала плотности (DFT), Естественные орбитальные связи и энергии для двух фторуглеродных соединений
Shahriar Ghammamy1, Ali Rahmani2*
1Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin, Iran
22Department of Chemistry, Faculty of Science, Azad University unit Of Takestan , Takestan, Iran
e-mail: shghamamiii@yahoo.com or Ali_Rahmani870@yahoo.com
Аннотация. Методом DFT ( Теория функционала плотности) c помощью B3LYP/6 расчетов были определены отдельные длины связей и валентные углы для C2Cl3H и C2Cl3F. Ключевая группа, характерная для обоих соединений - это C1.
Ключевые слова: Электронная структура, Фторуглеродные соединения, Теория функционала плотности, Колебательный анализ, B3LYP уровень.
Введение
Фторированные алканы имеют много областей применения. В нормальных условиях фторуглероды - это бесцветные жидкости с высокой плотностью почти вдвое большей, чем у воды. Благодаря слабым межмолекулярным связям эти соединения обладают низкой вязкостью в сравнении с другими жидкостями с такой же температурой кипения.
Фторуглероды – это фторорганическими соединениями, состоящие только из углерода и фтора. Фторалканы химически и термически более стабильны, чем соответсвующие алканы. Однако, фторуглероды с двойными (фторалкены) и особенно с тройными (фторалкины) связями более реакционноспособны, чем соответствующие им углеводороды.
Они не смешиваются с большинством органических растворителей (таких как этанол, ацетон, этил ацетат и хлороформ), но могут смешиваться с некоторыми углеводородами (такими как гексан в некоторых случаях). Они обладают очень низкой растворимостью в воде, также как и вода практически не растворяется в них (порядок 10 частей на миллион). Количество атомов углерода в молекуле фторуглерода во многом определяет физические свойства. Чем больше число атомов углерода, тем выше точка кипения, плотность, вязкость, поверхностное натяжение, критические свойства, упругость пара и индекс преломления.
Растворимость газа уменьшается по мере увеличения количества атомов углерода, а вот температура плавления определяется другими факторами и поэтому ее нелегко предсказать (1-6).
Фторалканы очень стабильны благодаря прочности и природе связи углерод-фтор. Они являются самыми прочными связями в органической химии. Их прочность — результат электроотрицательности фтора.
В настоящей работе авторами были изучены структуры и характеристики соединений фтора: C2Cl3H и C2Cl3F. Структура соединения была оптимизирована применением метода DFT (B3LYP) (теория функционала плотности) с 6-311 основным набором с использованием программы Gaussian 09w. Метод теории функционала плотности был применен для определения оптимальной структуры C2Cl3H и C2Cl3F. Первоначальные вычисления были произведены на уровне теории функциональной плотности и были использованы сплит-валентные положительной поляризации на базе 6-311 наборов. Местные минимумы были получены путем полной геометрической оптимизации, и имели все положительные частоты.
Экспериментальная часть
Оптимизированные структурные параметры были использованы в расчетах колебательных частот на уровне теории функциональной плотности, чтобы иметь возможность охарактеризовать все неподвижные точки как минимум. Все вычисления были произведены с помощью программы Gaussian 09w. Гармоничные частоты колебаний (ν) даны в см-1 , интенсивности инфракрасных спектров представлены в километре на моль для всех соединений. Энергетический минимум геометрии молекул определен минимизацией энергии по отношению ко всем геометрическим координатам без наложения симметричных ограничений.
Изучение NBO (Natural Bond Orbital, естественных орбитальных связей) на структурах
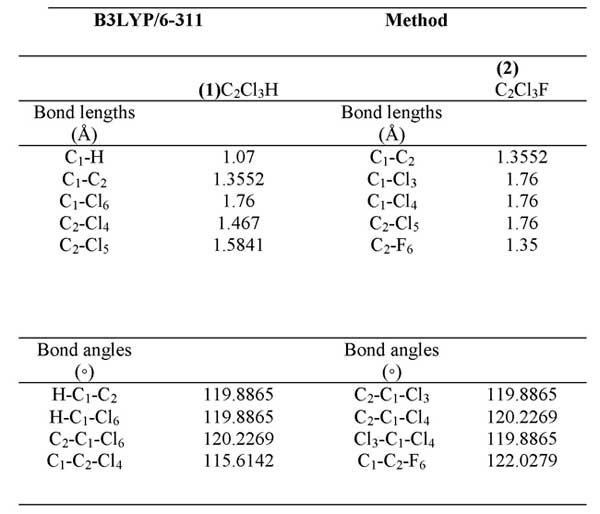
Расчет гибридизации NBO является важным параметром в нашем исследовании. Структура соединения была оптимизирована методом DFT с применением 6-311 основных наборов, с использованием программы Gaussian 09w. Метод теории функциональной плотности был использован для определения оптимальных структур C2Cl3H и C2Cl3F. (таблица 1, Рис. 1).

Рис. 1.
Таблица 1. Геометрические параметры, оптимизированные для (1)C2Cl3H, (2)C2Cl3F некоторых длин связей (Å) и углов (◦).
Естественные орбитальные связи локализованы на орбиталях с несколькими атомными центрами, которые описывают модель молекулярных, похожих на модель Льюиса, связей электронных пар в оптимально компактной форме. Точнее, естественные орбитальные связи это ортонормированный набор локализованных орбиталей «максимальной вместимости», чьи ведущие N/2 звенья (или N звенья в случае открытой оболочки) дают наиболее точное, возможно подобное Льюисовскому, описание полной электронной плотности. Этот вывод был сделан в следствие изучения всех возможных взаимодействий между «наполненных» (донорских) Льюисовского типа естественных орбитальных связей и «пустых» (акцептантов) не Льюисовского типа естественных орбитальных связей, и оценки их энергетических значений с помощью теории 2-го порядка возмущения.
Поскольку эти взаимодействия стремятся к передаче занятого местоположения из локализованных естественных орбитальных связей идеальной структуры Льюиса в пустые не-льюисовские орбитали. (и таким образом уход из идеальной структуры Льюиса). И они классифицируются как «делокализованные» поправки к нулевому порядку естественной структуры Льюиса. Естественные заряды были вычислены с использованием модуля NBO, реализованного в программе Gaussian 09w. Эти величины выводятся из анализа множества естественных орбитальных связей. Модель представляет собой орбитальную картину, которая ближе к классической структуре Льюиса. Анализ естественных орбитальных связей, включая скрещивание выбранных связей, представляется в методах B3LYP и 6-311 уровне теории (таблицы 2,3).
Таблица 2. NBO вычисленные гибридизации (1)C2Cl3H, (2) C2Cl3F при B3LYP/6-311.

Таблица 3.
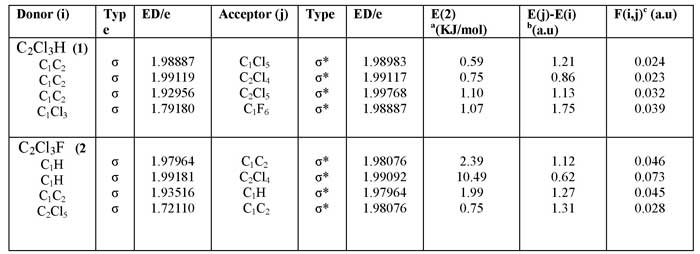
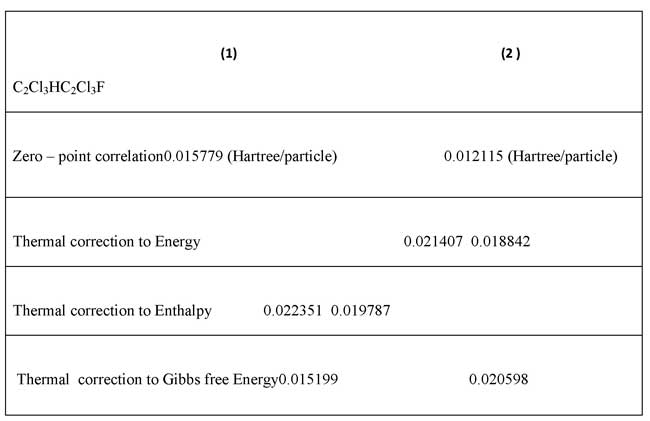
Эти данные показывают гипер сопряжение электронов между лигандами атомов с центральным атомом. Эти сопряжения сохраняют основу p-d π-связей. Расчитанные NBO гибридизации для C2Cl3H и C2Cl3F показывают, что все связи имеют SPX скрещивание и неплоские конфигурации. Полное скрещивание этих молекул является SPX , что подтверждается структурой. Количество связей гибридизации обнаруживает внутреннее равенство между центральными углами атомов (таблица 2) обнаруживая отклонения от нормальной стрктуры VSEPR и подтверждая отклонения от VSEPR структуры (фиг.2). Некоторые термодинамические параметры Частоты для (1)C2Cl3H, (2)C2Cl3F, энергии нулевой точки, корректирующая энергия, Энтальпия длины, свободная энергия Гиббса вычеслены и подтверждены в других теоретических работах (таб 4).
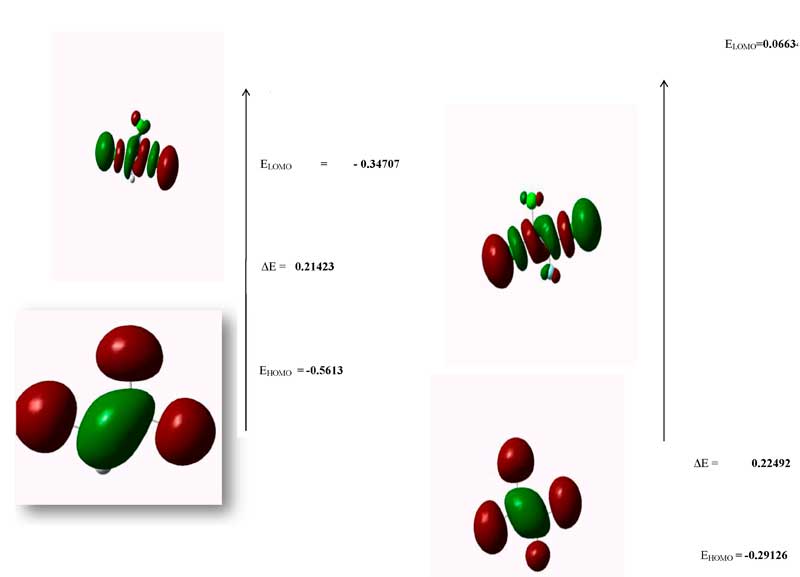
Рисунок 2.
Таблица 4. Некоторые термодинамические параметры для (1)C2Cl3H, (2) C2Cl3F
Теория граничных орбиталей (Frontier molecular orbital)
У HOMO (highest occupied molecular orbital) есть возможность передавать электрон, а у LUMO (lowest unoccupied molecular orbital) как у акцептанта есть возможность принимать электрон. Энергия HOMO и LUMO была вычислена методом B3LYP/6-311.Это электронное поглощение соответствует перемещению с основания к первому состоянию возбуждения и обычно описывается через возбуждение одного электрона с верхней занятой молекулы или орбитали (LUMO), а верхняя занятая молекулярная орбиталь и нижняя незанятая молекулярная орбиталь (LUMO) являются основными орбиталями участвующими в химической стабилизации. Поэтому, пока энергия HOMO напрямую связана с потенциалом ионизации, энергия LUMO напрямую связана со свойствами электрона. Энергетическая разница между HOMO и LUMO орбиталями называется энергетическим разрывом, что является важной для стабильностьи структур. В дополнение, 3D графики высших занятых молекулярных орбиталей (LUMO) представлены в фигуре 2. Расчет HOMO-LUMO энергии проведен также в 6-311 и величины приведены на Рис. 2 соответственно.
Выводы
В выполненной работе по двум фторуглеродным соединениям были произведены теоретические исследовании. Оптимизированные геометрии и частотные показатели стационарных точек и переходных состояний были рассчитаны с помощью
DFT(B3LYP
) методов с 6-311 основными наборами. Результаты расчетов показали, что некоторые выбранные величины длин связей и углов связей оцениваются для C2Cl3H
и C2Cl3F. Ключевая группа, характерная для обоих соединений - это C1.
Благодарности
Авторы выражают благодарность Научному Совету Международного университета Имама Хомейни за финансовую поддержку в виде гранта # 751387-91, а также Университет Tarbiat Modaress за техническую поддержку .
Литература
- O'Hagan, D . February 2008. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 37 (2): 308–19
- Sudha. S,Sundaraganesan.N,Kurt,Cinar.M. 2011 Karabacak, Molecularstructure, vibrational spectroscopic, first order hyperpolarizability and HOMO, LUMO studies of 2-aminobenzimidazole.Journal of Molecular Structure,84: 184-195.
- Lemal, D. M. 2004. Perspective on fluorocarbon c?hemistry. Journal of Organic Chemistry, 69 (1): 1–11.
- Murphy, C.D, Schaffrath, C. O'Hagan D2003Fluorinated natural products: the biosynthesis of fluoroacetate and 4-fluorothreonine in Streptomyces cattleya" Chemosphere,52(2):455-61.
- Lewandowski ,G. Meissner E, Milchert E.Hazard ,J. 2006. Special applications of fluorinated organic compounds.136(3):385–91.
- Becke, A. D. 1993 Density-Functional Thermochemistry. III. The Role of Exact
Exchange. J. Chem. Phys., 98: 5648-5652 - Sundaraganesan, N. and S. Ilakiamani, 2007. Dominic Joshua B Vibrational spectroscopy investigation using ab initio and density functional theory analysis on the structure of 3, 4-dimethylbenzaldehyde. Spectrochimica Acta Part A., 68: 680-687
- Lewis, D. F. V., C. Ioannides, and Parke, D. V. 1994. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: computer analysis of structure- activity relationships. Xenobiotica, 24: 401-408.
- Ralph, G. 1992. Chemical hardness and the electronic chemical potential Inorganic, chimica Acta, 198: 781-786.
- Zhang, W.Curran D.P. 2006. Synthetic Application of Fluorous. Tetrahedron 62: 11837–11865.
- Smith, M. C., Ciao.Y, Wang ,H. andGeorge, S. J. 2005. Coucouvanis D., Koutmos M, Sturhahn W, Alp EA, Zhao J, Kramer SP Normal-Mode Analysis of FeCl4- and Fe2S2Cl42- via Vibrational Mossbauer, Resonance Raman, and FT-IR Spectroscopies.Inorg. Chem., 44: 5562-5570.
- Vrajmasu, V. V., Mu¨nck,E. and E. L. Bominaar, 2004. Theoretical Analysis of the Jahn−Teller Distortions in Tetrathiolato Iron(II) Complexes. Inorg. Chem., 43: 4862–4866.
- Ghammamy, Sh., K. Mehrani,Rostamzadehmansor, S. and Sahebalzamani,H. 2011. Density functional theory studies on the structure, vibrational spectra of three new tetrahalogenoferrate (III) complexes. Natural Science, 3, 683-688.
- Frisch, M. J. Trucks, G. W. 1998. GASSIAN 98 (Revision A. 3) Gaussian Inc., Pittsburgh, PA, USA.
Материал рекомендован к публикации членом редколлегии С.М. Игумновым
Fluorine Notes, 2013, 86, 7-8
