Received: May, 2012
Fluorine Notes, 2013, 86, 3-4
SYNTHESIS, STRUCTURE, AND COMPOSITION OF FLUORINATED FLUOROPOLYMER PRODUCTS OF REACTIONS BETWEEN 4.4'-DIPHENYLMETHANE DIISOCYANATE AND 1.1.5-TRIHYDROPERFLUOROPENTANOL-1
S. V. Kudashev (corresponding author), N. A. Rakhimova, V. F. Zheltobryuhov, V. N. Arisova
Volgograd State Technical University Russian Federation, 400005, Volgograd, Lenin ave., 28
e-mail:
kudashev-sv@yandex.ru
Abstract:The reaction between 4.4'-diphenylmethane diisocyanate and 1.1.5-trihydroperfluoro-pentanol-1 resulted in the formation of fluorinated urethanes. The structure of the synthesized products was studied by the method of X-ray diffractometry, IR, and NMR (1H,13C,15N,19F) spectroscopy. The impact of the solvent polarity both on the conversion of 1,1,5-trihydroperfluoropentanol-1, and on the composition of products resulting from the urethane formation is demonstrated.
Keywords: 4.4'-diphenylmethane diisocyanate, polyfluorinated alcohol, urethane, tin di-n-butyldilaurate.
The development of valuable useable materials with improved properties is an urgent challenge in the applied polymer chemistry. Therefore, the polymers stabilized by polyfluorinated or perfluorinated substances are known to have increased indices of hydrolytic stability, light-, thermo-, and wear-resistance, and lower flammability as well [1, 2].
It is of interest to undertake the synthesis of novel reactive poly- or perfluorinated substances targeted for use as modifiers in macromolecular systems. As a method for carbamates (urethanes) manufacture the process of nucleophilic addition is widely applied not only in the synthesis of polymers, but as their modifier as well [3-6]. Though the reactions of aromatic diisocyanates with substances containing «active» hydrogen (alcohols, carbonic acids, amines) were studied sufficiently, the application of those substances with electron-withdrawing substituents presents definite problems associated with their –I-effects, and consequently decreased reactivity [5, 6].
The objective of this study was to investigate both the compositions and structures of products of the interaction of 4.4/-diphenylmethane diisocyanate (MDI) with 1.1.5-trihydroperfluoropentanol-1 (PFS2) at the conditions of its catalysis by tin di-n-butyldilaurate (DBDLO) in organic solvents with variable dielectric permeabilities.
Discussion of results
The interaction between MDI and PFS2 at their 1:1 mole-to-mole ratio leads to a mixture of fluorinated polymer products: mono- (I) and diurethane (II), and to such cyclization products as 1.4-disubstituted uretidinedione (III) and 1,3,5-trisustituted isocyanurate (IV):
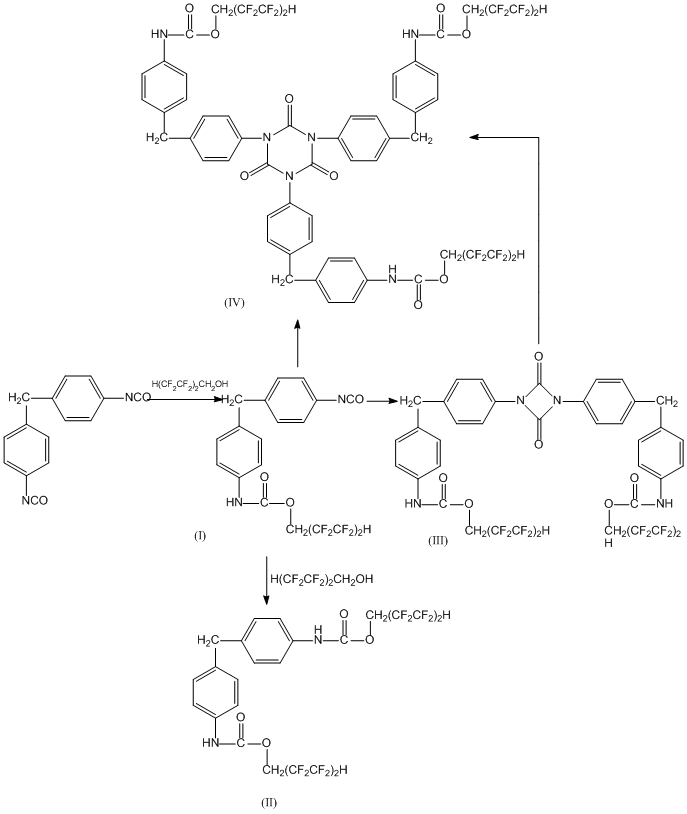
The degree of alcohol conversion grows with the solvent polarity and the reaction temperature (see Table). One may see that the introduction of an additional chlorine atom into the aromatic ring influences strongly not only the degree of alcohol transformation, but also the composition of the prepolymer reaction products. At the same time in more polar o-dichlorobenzene the share of difficultly separable product blend (V) grows considerably to 21% mass, while that of cyclic prepolymers (III) and (IV) goes down accordingly.
Table – The influence of solvent polarity and temperature on the degree of alcohol conversion and the composition of reaction products
|
Solvent |
Dielectric permeability, Оµ (25 В°C) |
Temperature of synthesis, В°C |
Degree of conversion PFS2, % |
Composition of the reaction products, % mass |
|||
|
I |
II + III |
IV |
V* |
||||
|
В Chlorobenzene |
В 5.6 |
60 |
55.4 |
60 |
21 |
12 |
7 |
|
80 |
76.2 |
78 |
12 |
5 |
5 |
||
|
100 |
78.1 |
78 |
15 |
3 |
4 |
||
|
o-dichlorobenzene |
В 9.9 |
60 |
57.2 |
67 |
17 |
8 |
8 |
|
80 |
82.6 |
65 |
17 |
3 |
15 |
||
|
100 |
84.7 |
68 |
7 |
4 |
21 |
||
* difficultly separable product blend.
IR-Fourier spectral study of the difficultly separable product blend (V) demonstrated intensive adsorption bands at 1715-1770 cm-1, here assigned to the valent vibrations of carbonyl-groups in urethane, allophanate, uretidinedione, and isocyanurate fragments [3, 7]. It is also remarkable that the wideness and position of HN-group adsorption band at 3421-3545 cm-1 are due not only to the >C=Oв€™в€™в€™HN~ interactions but also to the association between proton-donating fragments and fluorines belonging to the perfluorinated chain, their presence in product (V) composition being revealed by characteristic C-F vibration bands at 1120.4-1145.4 cm-1. Low solubility of the said product blend in organic solvents at 100 В°C (benzene, toluene, nitromethane) points to the presence of cross-linked structures formed via the reaction between isocyanate groups and urethane fragments.
It is typical for NMR 1H spectra of product (V) that the proton signal of difluoromethyl group belonging to perfluorocarbon chain with related spin-spin interaction constant 5.77 t.t (-CF2H, J1 = 51.9 Hz, J2 = 5.7 Hz), and that the methylene fragment proton 4.55 t (-CH2-CF2, J = 14.1 Hz) are present simultaneously. In their turn NMR 13C spectroscopy results show the signals assigned to the carbon of urethane group (165.13 ppm), carbonyl of 1,4-disubstituted uretidinedione cycle (154.78 ppm), and carbonyl of 1,3,5-trisubstituted isocyanurate cycle (157.56 ppm). Our analysis of signals in NMR 15N spectra leads to the conclusion about the presence of urethane (-202.2 ppm), allophanate (-30.7 ppm) groups, and some cyclization products (-36.4 and -38.5 ppm).
Our studies on the structure of prepolymer (III) and (IV) blend by the method of X-ray analysis revealed a complicated diffraction picture containing numerous reflects due to the products crystal structure (Figure 2). Considerable input into the intensity of those reflects and their angle position is contributed by the equilibrium process of transformation of the four-member nitrogen-containing heterocycle to more thermodynamically stable six-member one, manifested by the reflects intensity increase. Our comparative analysis of experimental diffractograms at wide diffusion angles points at the occurrence of distant reflects 2Оё = 52В° (d = 1.82 Г…), and 2Оё = 54В° (d = 1.70 Г…), brought about by the shift of those molecules along their axes, their hades and bending during the heterocycle conformation inversion [8].
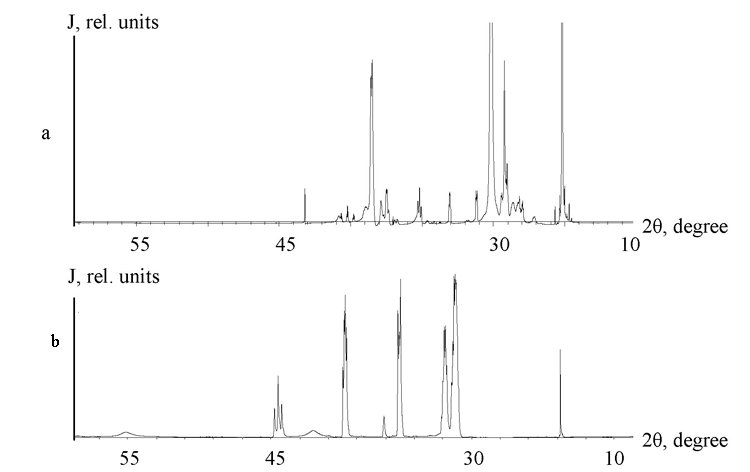
Fig. 1. Diffractogram of cyclic prepolymer (III) and (IV) blend. a – fresh sample; b – after 24 hours; J – relative intensity; 2θ – diffraction angle.
The application of the techniques of two-dimensional proton NMR-spectroscopy and IR-Fourier spectrometry made it possible to specify that for prepolymers (III + IV) in chlorobenzene or o-dichlorobenzene solutions at temperatures ≥50 °C partial decyclization is typical followed by the generation of isocyanate groups, that is manifested through the decline in intensity and position alteration of the proton -NH-CO- (7.08 ppm, marked*) cross-peak, and the appearance at 2277 cm-1of an intensive band assigned to the isocyanate group valent vibrations (Fig.2 and Fig.3) initially not present in crystal forms (III + IV). The form of diagonal cross-peaks in 2D NMR-spectra evidences of associative interactions between proton-donative and proton-accepting groups in prepolymer molecules.
When the solvent polarity increases the change both in the form and position of -NH-CO-proton diagonal cross-peak is due to the opening of uretidinedione cycle and related decrease in the share of associative interactions >C=O в€™в€™в€™ HN~. The said fact is in good conformity with the results of NMR-spectroscopy research with 15N and 19F nuclei. Thus, it was found that when prepolymer blend (III + IV) is dissolved in chlorobenzene at 70 В°C, in its NMR 15N spectra the signals observed at 36.1 ppm and assigned to nitrogen of the four-member uretidinedione cycle disappear, while the characteristic peak of nitrogen of six-member isocyanurate heterocycle (38.5 ppm) stays unchanged. At the same time, however, the opening of cycle of prepolymer (III) in fact provides no effect on the chemical shift values in NMR 19F spectra: -33.6Г·40.5 ppm (-CH2CF2-), -45.9Г·50.5 ppm (-CF2-CF2H), -57.9 ppm (-CF2-) (Fig. 4).

Fig.2. IR-Fourier spectrum of prepolymer (III + IV) blend dissolved in o-dichlorobenzene at 70 В°C.
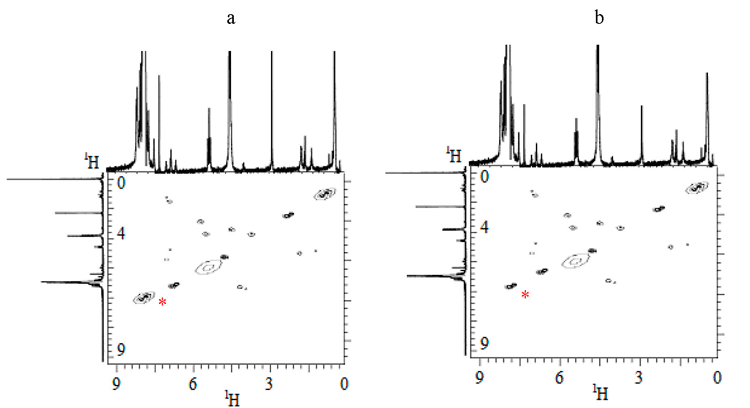
Fig. 3. 2D-COSY 1H-1H NMR-spectrum of prepolymer (III + IV) blend dissolved in chlorobenzene (a) or in o-dichlorobenzene (b) at 70 В°C.
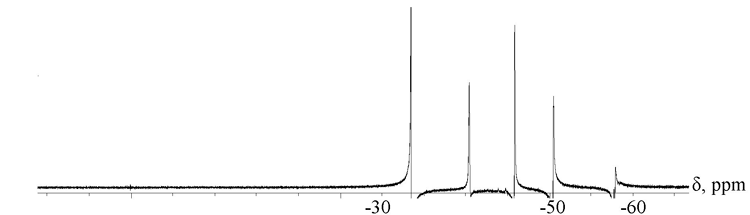
Fig. 4. 19F NMR-spectrum of prepolymer (III + IV) blend dissolved in o-dichlorobenzene at 70 В°C.
Therefore, we conducted the synthesis, determined molecular and crystal structures and composition of the products of the reaction between equimolar quantities of 4.4'-diphenylmethane diisocyanate and 1.1.5-trihydroperfluoropentanol-1, and demonstrated the formation not only of mono- and diurethane, but also such cycled products as fluorinated substituted uretidinediones and isocyanurates. It is proved that the increase in the solvent polarity favors the alcohol conversion, and partial decyclization of prepolymers followed by the generation of isocyanate groups.
Experimental
In our urethane synthesis we used industrially produced 4.4'-diphenylmethane diisocyanate and 1.1.5-trihydroperfluoropentanol-1; their interaction was conducted in chlorobenzene or o-dichlorobenzene environment (the solvent purity qualification was В«Pure for analysisВ»). Tin di-n-butyldilaurate in the form of 0.5% mass. solution in chlorobenzene or o-dichlorobenzene was used for the catalyst of urethane formation.
Pattern of the synthesis technique. 40 ml of solvent, 1g (0.004 mole) of 4.4'-diphenylmethane diisocyanate, 0.6ml (0.004 mole) of 1,1,5-trihydroperfluoropentanol-1 were charged into a glass bulb equipped with a reflux condenser provided with a calcium chloride tube, and 0.1 ml of tin di-n-butyldilaurate was added in the form of 0.5 % mass. solution in chlorobenzene. The bulb was being thermostated at 80 В°C during 2 hours being exposed to ultrasound 40 kHz. The reaction products then formed were fractionized: prepolymer (I) is soluble in chlorobenzene at 25 В°C, prepolymer (II) is soluble in chlorobenzene at 80 В°C, prepolymers (III and IV) are poorly soluble in chlorobenzene at 25 В°C. The products were washed with n-hexane, dried over waterless calcium chloride, and stored in sealed ampoules.
IR-spectra were recorded with the help of IR-Fourier spectrometer В«Nicolet-6700В» or spectrometer В«Specord-M82В». NMR-spectra (1H, 13C, 15N, 19F) were registered with the help of В«Varian Mercury PlusВ» (operative frequency was 300 MHz) in DMSO-d6 at 25 В°C. 1H and 13C chemical shifts were recorded with reference to tetramethylsilane (TMS); the measurement accuracy was 0.01 and 0.02 ppm correspondingly.
15N and 19F chemical shifts were recorded with accuracy 0.1Hz with reference to internal standard (nitromethane or fluorobenzene, correspondingly). The technique of two-dimensional homonuclear 2D-COSY NMR 1H–1H spectroscopy was applied to assign signals and determine the substance structure.
The prepolymer crystal structure was studied by the method of X-ray analysis, using the standard technique, with the help of automatic diffractometer DRON-3, CuKО± (О» = 1.5418 Г…) radiation, graphitic monochromator using the secondary ray beam, the generator regime was 36kVx20mA. Fit2D software was used for experimental diffractogram processing.
Prepolymer (I). Ochroleucous crystals. Determined: N 5,76-5,93 %, F 29,13-33,21. Calculated: N 5,81 %, F 31,51. IR-spectrum, ОЅ, cm-1: 3196-3076 (ОЅN-H), 2914-2986 (ОЅC-H), 2338 (ОЅNCO), 1834 (ОЅC=O, carbamine), 1684-1570 (Car-Car), 1666 (amide I), 1552 (amide II), 1462 (amide III), 1168-1198 (C-F). Spectrum NMR 1H (CCl4), Оґ, m. d.: 6,39-7,22 m (superposition of 8H, C6H4 , and 1H, C(O)NH signals), 5,79 t.t (1H, -CF2H, J1 = 51,9 Hz, J2 = 5,7 Hz), 4,57 t (2H, CH2-CF2, J = 14,1 Hz), 3,83 t (2H, C6H4-CH2-C6H4, J = 6,6 Hz). Spectrum NMR 13C (DMSO-d6), Оґ, m. d.: 157,20 (1C, >C=O, carbamine), 117,48-136,62 (superposition of 12C, C6H4 and 4C, CF2), 127,87 (1C, NCO), 37,60-39,26 (1C, C6H4-CH2-C6H4).
Prepolymer (II). Ochroleucous crystals. Determined: N 3,71-3,95 %, F 40,06-43,01. Calculated: N 3,92 %, F 42,55. IR-spectrum, ОЅ, cm-1: 3308-3106 (ОЅN-H), 2910-2917 (ОЅC-H), 1835 (ОЅC=O, carbamine), 1674-1582 (Car-Car),1661 (amide I), 1559 (amide II), 1468 (amide III), 1217-1109 (C-F). Spectrum NMR 1H (CCl4), Оґ, m. d.: 6,37-7,20 Рј (superposition of signals 8H, C6H4 and 2H, C(O)NH), 5,77 t.t (2H, -CF2H, J1 = 51,9 Hz, J2 = 5,7 Hz), 4,55 t (4H, CH2-CF2, J = 14,1 Hz), 3,84 t (2H, C6H4-CH2-C6H4, J = 6,6 Hz). Spectrum NMR 13C (DMSO-d6), Оґ, m. d.: 154,80 (1C, >C=O, carbamine), 117,48-136,62 (superposition 12C, C6H4 and 8C, CF2), 37,68-39,22 (1C, C6H4-CH2-C6H4).
Prepolymers (III+IV). Ochroleucous crystals. Determined: N 5,71-6,02 %, F 30,08-34,33. Calculated: N 5,81 %, F 31,51. IR-spectrum, ОЅ, cm-1: 3319-3158 (ОЅN-H), 2920-2993 (ОЅC-H), 1894 (ОЅC=O, carbamine), 1884-1881 (ОЅC=O, isocyanurate and uretidinedione), 1683-1571 (Car-Car),1665-1670 (amide I), 1557-1564 (amide II), 1463-1482 (amide III), 1215-1101 (C-F). Spectrum NMR 1H (CCl4), Оґ, m. d.: 6,37-7,28 m (superposition of signals C6H4 and C(O)NH), 5,81 t.t (-CF2H, J1 = 51,9 Hz, J2 = 5,7 Hz), 4,52 t (CH2-CF2, J = 14,3 Hz), 3,81 t (C6H4-CH2-C6H4, J = 6,6 Hz). Spectrum NMR 13C (DMSO-d6), Оґ, m. d.: 161,80 (>C=O carbamine), 157,20 (>C=O, isocyanurate and uretidinedione), 117,48-136,62 (superposition of C6H4 and CF2 signals), 37,60-39,26 (C6H4-CH2-C6H4).
References
- Sintez, struktura, svojstva i tribotekhnicheskie harakteristiki materialov na osnove politiouretanovyh kompozicij, modificirovannyh poliftor- i med'soderzhashchim Na+-montmorillonitom v usloviyah termookislitel'nogo i svetovogo stareniya / I.A. Novakov, N.A. Rahimova, A.V. Nistratov, S.V. Kudashev, S.Yu. Gugina // Trenie i iznos. - 2011. - T. 32, N 5. -s. 476-488.
- Buznik, V. M. Sostoyanie otechestvennoj himii ftorpolimerov i vozmozhnye perspektivy razvitiya / V. M. Buznik // Ros.khim. zh.(Zh. Ros. Khim. ob-va im. D. I. Mendeleeva).-2008.-T.LII, N 3.-s. 7-12.
- Tiger, R. P. Polimerizaciya izocianatov / R. P. Tiger, L. I. Sarynina, S. G. Entelis // Uspekhi khimii. – T. XLI. – Vyp. 9. – 1972. – s. 1672-1695.
- Entelis, S. G. Kinetika i mekhanizm reakcij izocianatov s soedineniyami, soderzhashchimi «aktivnyj» vodorod / S. G. EHntelis, O. V. Nesterov // Uspekhi khimii. – T. XXXV. – Vyp. 12. – 1966. – s. 2178-2203.
- Mashlyakovskij, L. N. Sintez makrodiizocianatov na osnove ftorirovannyh diolov. CH. 1 Kinetika vzaimodejstviya ftorirovannyh diolov s cikloalifaticheskimi i aromaticheskimi diizocianatami / L. N. Mashlyakovskij, E. V. Homko, K. Tonelli // Lakokrasochnye materialy i ih primenenie. – 2002. – N4. – s.8-16.
- Rahimova, N. A. Osobennosti reakcii poliizocianata s poliftorirovannymi spirtami / N. A. Rahimova, S. V. Kudashev // Izv. VolgGTU. Seriya «Khimiya i tekhnologiya ehlementoorganicheskih monomerov i polimernyh materialov». Vyp. 8: mezhvuz. sb. nauch. st. / VolgGTU. - Volgograd, 2011. - N 2. - s. 133-140.
- Bellami, L. Infrakrasnye spektry slozhnyh molekul / L. Bellami. – M.: Inostrannaya literatura.-1963.-345 s.
- Vekilova, G. V. Difrakcionnye i mikroskopicheskie metody i pribory dlya analiza nanochastic i nanomaterialov / G. V. Vekilova, A. N. Ivanov, Yu. D. Yagodkin. – M.: MISiS. – 2009. – 145 s.
Recommended for publication by Prof. Alexander I. Rakhimov
Fluorine Notes, 2013, 86, 3-4
