Received: : ноябрь 2012
Fluorine Notes, 2012, 85, 5-6
СИНТЕЗ И ИЗУЧЕНИЕ РЕАКЦИОННОЙ СПОСОБНОСТИ ПЕРФТОРБЕНЗИЛКЕТОНОВ
Г.А.Емельянова, В.В.Беренблита, С.Р.Стерлинб
а ФГУП Научно-исследовательский институт синтетического каучука им. ак. С.В.Лебедева,
198035 Санкт-Петербург, ул. Гапсальская, 1
e-mail: emelianovgennadiy@yandex.ru
б Институт элементорганических соединений им. ак. А.Н.Несмеянова РАН, Москва, ул. Вавилова, 28
Аннотация. Жидкофазным окислением перфтораллилбензола получен 1,2-эпоксиперфтор-3-фенилпропан, который был превращен в перфторметилбензилкетон и фторсульфонилоксидифторметилперфторбензилкетон под действием SbF5 или HSO3F соответственно. Изучены методы синтеза перфторированных виниловых эфиров на основе синтезированных кетонов.
Ключевые слова: 1,2-эпоксиперфтор-3-фенилпропан, перфторметилбензилкетон, фторсульфонилоксидифторметилперфторбензилкетон.
Уникальная хемо- и термостойкость перфторированных каучуков насыщенного ряда определяется высоким - 107÷121 ккал/мол - значением энергии C-F-связи [1]. В то же время именно это свойство фторорганических соединений не позволяет осуществить вулканизацию перфторированных каучуков методами, традиционно используемыми при вулканизации нефторированных или частично фторированных каучуков [2].
Одним из решений этой задачи является получение перфторкаучуков с участием мономеров, содержащих функциональную группу, способную взаимодействовать с нуклеофильными реагентами. Так, сополимер перфторфенилглицидилового эфира с ОГФП вулканизуется под действием дикалиевой соли бис-фенола А (атмосфера N2, 200 °C/3 дня) [3].
Можно было ожидать, что в перфторированных мономерах жирноароматического ряда наличие фторалкильного заместителя будет повышать электрофильность пентафторфенильной группы, и вулканизация каучуков, содержащих такие мономеры, под действием дифункциональных нуклеофилов будет осуществляться в более мягких условиях.
С целью получения перфторвиниловых эфиров, содержащих перфторбензильную группу, нами была изучена возможность получения перфторированных бензилкетонов (4, 6).
Кетоны 4 и 6 синтезированы по схеме, включающей в себя жидкофазное окисление перфтораллилбензола (1)[4]:
Схема 1

и электрофильную изомеризацию образующейся окиси перфтораллилбензола (2) под действием SbF5, приводящую к образованию кетона 4 (схема 2), или HSO3F с образованием кетона 6 (схема 3).
В присутствии каталитических количеств SbF5 (мольное соотношение 1: SbF5 = 60:1) окись 2 в результате экзотермической реакции образует смесь 4 и перфтортолуола в мольном соотношении 9:1:
Схема 2
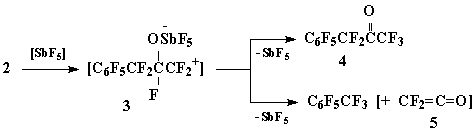
Учитывая ничтожное количество взятой в реакцию SbF5 образование перфтортолуола можно объяснить схемой распада промежуточного иона карбения 3, включающей в себя элиминирование стабильного перфторбензильного катиона с одновременным отрывом аниона фтора из α-положения и генерацией дифторкетена (5).
Реакция окиси 2 с HSO3F, приводящая к образованию фторсульфонилокси-кетона 6, осуществляется в относительно мягких условиях (100 оС/1,5 часа); аналогичная реакция ОГФП требует нагревания до 200÷ 220 оС [5]
Схема 3
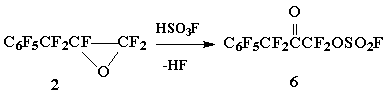
Подобно другим перфторированным кетонам 4 образует водорастворимый гидрат, а в присутствии каталитических количеств CsF при 0÷ 15 оС гладко присоединяется к ОГФП, давая моноаддукт (7). В то же время в более жестких условиях - 80÷ 85 оС - при катализе CsF кетон 4 претерпевает внутримолекулярную циклизацию, давая перфтор-2-метилбезофуран (8):
Схема 4

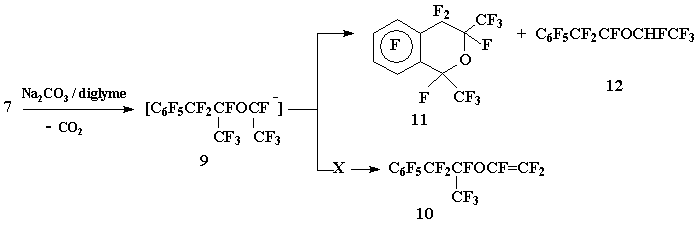
Попытка получить трифторвиниловый эфир (10) дефосгенированием фторангидрида 7 действием Na2CO3 в диглиме привела к образованию перфтор-1,3-диметил-2-хромана (11) в качестве основного продукта реакции в результате нуклеофильного замещения атома фтора в о-положении ароматического ядра карбанионом (9), генерируемым в ходе реакции, и β-(1,2,2,2-тетрафторэтокси) перфторпропилбензола (12):
Схема 5
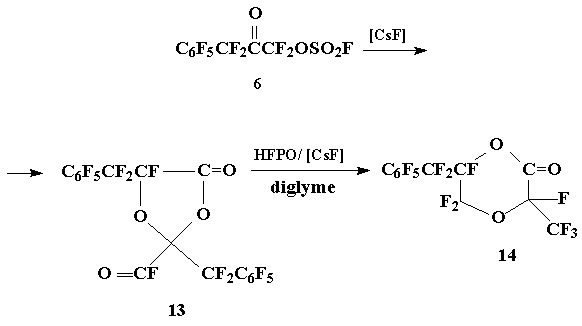
Альтернативным методом получения фторолефина на основе окиси 2 могла бы явиться схема, аналогичная схеме синтеза перфтор-2-метилен-4-метил-1,3-диоксолана [6 а÷в], включающая в себя превращение кетона 6 в димер фторангидрида α-кетокислоты (13), получение циклоаддукта (13) с ОГФП, изомеризацию образующегося 2-оксо-1,4-диоксана (14) в присутствии CsF в 2-фторформил-1,3-диоксолан (15) и омыление (15) водным раствором Na2CO3 с последующим пиролизом образующегося карбоксилата .
4-Оксо-1,3-диоксолан (13) был получен разложением фторсульфонилоксикетона 6 под действием CsF; последующая реакция (13) с ОГФП/[CsF] приводит к образованию 2-оксо-1,4-диоксана (14) (об образовании гомодимеров перфторированных α-оксоалканоилфторидов и их циклоаддуктов с ОГФП см. также [7÷ 9]):
Схема 6
Однако изомеризация диоксана (14) в условиях, описанных в [6], привела к образованию неразделяемой ректификацией смеси ожидаемого 2-фторформил-1,3-диоксолана (15) и перфтор-2-бензофурилметоксипропионилфторида (16) с преобладанием последнего [(16): (15) = 5:4]. Как и в вышеописанных примерах, нуклеофильное замещение атома фтора в ароматическом ядре оказывается доминирующим направлением:
Схема 7
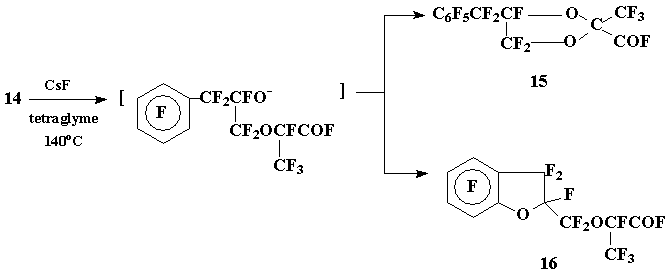
Обработкой смеси фторангидридов 15÷16 водным Na2CO3 и последующим пиролизом образовавшихся карбоксилатов натрия была получена смесь трифторвинилового эфира (17) и 2-метилен-1,3-диоксолана (18), которые были охарактеризованы спектрами ЯМР 19F:
Схема 8

Таким образом, проведенные исследования показали, что превращения производных перфторбензилкетонов с участием промежуточно генерируемых алкокси- или карбанионов, неизбежно сопровождаются образованием продуктов внутримолекулярного нуклеофильного замещения, по всей вероятности вследствие повышенной электрофильности ароматического ядра в составе перфторбензильной группы.
Экспериментальная часть
Спектры ЯМР 19F записаны на спектрометре Bruker AVANCE 300 при 282 МГц; внешний эталон CF3COOH. Масс-спектры записаны на спектрометрах VG ANALYTICAL 70-70E (70 eV) и Finnigan Polaris/GCQ Plus (70 eV).
Получение окиси перфтораллилбензола 2
В стальной реактор объемом 0,2 литра помещают 39 г (0,13 М) перфтораллилбензола и 150 мл хладона-113. После герметизации автоклав термостатируют в интервале 140÷150 оС и подают кислород до окончания поглощения. Ректификацией реакционной массы получают 29 г (70%) окиси 2, т.кип. 51÷ 52 оС/15 мм.рт.ст.. Найдено %: С 35,10; F 60,20. С9F10O. Вычислено %: С 34,40; F 60,50.
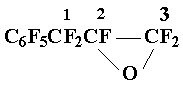
ЯМР19F : АВ-система c центрами 31,0 и 33,0 (2F3 ) ( JAB = 265 Гц ); 72,3 (1F2); АВ- система c центрами 20,0 и 28,1 (2F1); 60,7 (2 о-F) 68,3 (р-F), 81,5 (2 m-F).
Получение перфторметилбензилкетона 4
Окись 2 (56 г, 178 ммол) прибавляют по каплям к 0,7 г SbF5, поддерживая температуру реакционной массы не выше 40-45 оС, интенсивно перемешивают еще 10 мин; по данным ЯМР 19F смесь содержит 90% кетона 4 и 10% толуола 1. К реакционной массе прибавляют 4 мл безв. сульфолана, нагревают при перемешивании до 105÷110 оС, охлаждают, жидкую часть реакционной смеси декантируют со смолистого осадка, фильтруют и перегонкой выделяют 7 г фракции (т. кип. 125÷135 оС), содержащей смесь 4:C6F5CF3 = 1:1, и 42 г фракции, содержащей смесь 4:C6F5CF3 = 10:1; выход кетона 4 75%. Дальнейшей ректификацией выделен аналитический образец 4, т.кип. 141÷ 142,5оС. Найдено %: F 59,94. C9F10O. Вычислено %: F 60,51.
Спектр ЯМР 19F: — 1,3 (3F1); 23,8 (2F2); 63,0 (2 o-F); 72,0 (1 p-F); 85,0 (2 m-F).
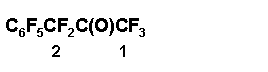
Получение фторсульфонилоксидифторметилгептафторбензилкетона 6
Смесь окиси 2 (13,3 г, 42 ммол) и HSO3F (8,5 г, 85 ммол) нагревают в стальной пробирке, защищенной от влаги воздуха, при 100 оС/1,5 часа, выливают на лед, органический слой промывают лед. водой, сушат SOCl2 (до прекращения выделения газообразных продуктов при температуре кипения SOCl2), избыток тионилхлорида отгоняют, из остатка перегонкой выделяют 9 г (52%) кетона 6, т.кип. 80÷ 83 оС/7÷ 8 мм.рт.ст.. Найдено %: C 28,10; F 47,86; S 7,73. C9F10O4S. Вычислено %: C 27,41; F 48,22; S 8,12.
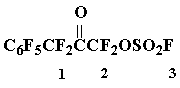
Спектр ЯМР 19F: —126,0 (1F3); 1,1 (2F2); 23,2 (2F1); 63,0 (2 o-F); 71,0 (1 p-F); 84,0 (2 m-F).
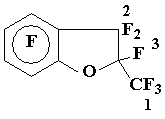
Получение ацилфторида 7
К смеси кетона 4 (9,5 г, 30 ммол), CsF (0,5 г) и 2 мл диглима при 0оС прибавляют ОГФП (5 г, 30 ммол), перемешивают при 0÷15 оС 15 мин, отогревают до ~25 °C, нижний слой (13,3 г) отделяют и перегонкой выделяют 11,4 г фракции (т.кип. 52÷74 оС/12 мм.рт.ст..), содержащей ~95% ацилфторида 8 (смесь стереоизомеров); выход 8 75%. Ректификацией выделяют аналитический образец, т.кип. 69÷ 74 оС/12 мм.рт.ст. (смесь диастереомеров). Найдено %: С 29,98; F 63,46. C12F16O2. Вычислено %: С 30,00; F 63,33. Спектр ЯМР 19F: —102,6 (1F1); 1,0 + 2,3 (3F4); 6,3 + 6,4 (3F2); группа сигналов 27,0÷ 32,0 (2F6); 48,8 + 51,5 (1F3); 61,7 (2 o-F); 65,0 (1F5); 72,0 (1 p–F); 85,5 (2 m-F).
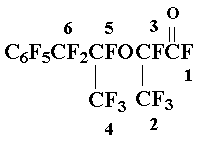
Получение бензофурана 8
К смеси CsF (0,7 г) и 4 мл тетраглима прибавляют по каплям кетон 4 (4,5 г, 14 ммол), по окончании экзотермической реакции полученный раствор нагревают до 80÷ 85оС до прекращения выпадения осадка; летучие при 70 оС/1÷2 мм.рт.ст.. продукты эвакуируют в приемник (-78 оС), полученный конденсат перегоняют и получают 3,9 г (86%) бензофурана 8, т.кип. 74÷76 оС/70 мм.рт.ст.. Найдено %: С 34,71; F 59,61. C9F10O. Вычислено %: С 34,39; F 60,51.
Спектр ЯМР 19F: 5,3 + 5,35 (3F1); AB-система с центрами 25,2 и 31,2, JA-B = 235 Гц (2 F2); 57,0 (1F3); 64,5 + 68,7 (2 o-F); 83,6 + 84,4 (2 m-F).
Реакция ацилфторида 7 с Na2CO3
К смеси Na2CO3 (1,2 г, 12 ммол) и 15 мл диглима при 65÷70 оС постепенно прибавляют ацилфторид 8 (5,4 г, 11 ммол), перемешивают при 140 оС до прекращения газовыделения, реакционную массу охлаждают, промывают разб. aq. HCl, органический слой (4 г) отделяют и перегонкой над H2SO4 выделяют 3,3 г смеси хромана 11 и гидрида 12 (11:12 = 4:1) (ХМС).
Масс-спектр 11 (m/z, отнесение): 414 [M]+ 20; 395 [M-F]+ 18; 367 [M-COF]+ 10; 345 [M-CF3]+ 100; 326 [C10F10O]+ 2; 317 [C9F11]+ 8; 295 [C9F9O]+ 2; 276 [C9F8O]+ 8; 267 [C9F7O]+ 18; 248 [C8F8]+ 15; 245 [C8F7O]+ 17; 229 [C8F7]+ 12; 226 [C8F6O]+ 8; 217 [C7F7]+ 6; 198 [C7F6]+ 30; 179 [C7F5]+ 20; 167 [C6F5]+ 2; 160 [C7F4]+ 4; 148 [C6F4]+ 5; 141 [C7F3]+ 5; 129 [C6F3]+ 3; 124 [C4F4]+ 5; 117 [C5F3]+ 5; 110 [C6F2]+ 2; 98 [C5F2]+ 2; 93 [C3F3]+ 5; 79 [C5F]+ 2; 69 [CF3]+ 31.
Масс-спектр 12 (m/z, отнесение): 434 [M]+ 7; 318 [C9HF11]+ 1; 267 [C9F7O]+ 5; 217 [C7F7]+ 100; 198 [C7F]+ 2; 167 [C6F5]+ 2; 101 [C2HF4]+ 10; 69 [CF3]+ 8; 51[CHF2]+ 3.
Получение 4-оксо-1,3-диоксолана 13 и 2-оксо-1,4-диоксана 14
К смеси CsF (0,8 г) и 3 мл моноглима прибавили по каплям кетон 6 (18,3 г, 46 ммол) и перемешивали до прекращения газовыделения. Образование диоксолана 13 (смесь изомеров) зафиксировано по данным ЯМР 19F: 27,0 + 29,0 (2 CF2); 41,2 + 44,2 (CF); 59,5 + 61,5 (4 o-F); 68,5 + 70,0 (2 p-F); 83,5 (4 m-F) (сигнал COF-группы в спектре не виден). Полученную смесь перенесли в стальной 50 мл автоклав, в автоклав переконденсировали ОГФП (8,3 г, 50 ммол), встряхивали при ~25 °C/48 час, перегонкой реакционной смеси получили 15 г (71%) диоксана 14 (смесь изомеров), т.кип. 64÷ 70 оС/2÷2,5 мм.рт.ст.. Найдено %: C 32,40; F 56,54. C12F14O3. Вычислено %: С 31,44; F 58,08.

Спектр ЯМР 19F: 24,0 + 30,0 (2F1); 46,8 + 48,0 (1F2); группа сигналов 4,0-16,9 (2F3 + 3F5); 35,0 + 39,2 (1F4); 61,5 + 63,0 (2 o-F); 70,8 + 72,0 (1 p-F); 85,0 (2 m-F).
Изомеризация 2-оксо-1,4-диоксана 14 под действием CsF
Смесь диоксана 14 (14,8 г, 32 ммол), CsF (0,4 г) и 2 мл тетраглима перемешивали при 140оС/2 часа, перегнали и получили 12,0 г смеси изомеров 15÷ 16 с т.кип. 41÷ 46 оС/1,5÷ 2 мм.рт.ст.. Найдено %: C 32,11; F 57,28. C12F14O3. Вычислено %: С 31,44; F 58,08. В масс-спектре обоих изомеров присутствует молекулярный ион 458 [M]+.
Получение винилового эфира 17 и олефина 18
Смесь фторангидридов 15÷ 16 (5,75 г) обработали aq. Na2CO3, водный раствор экстрагировали эфиром, эфир упарили, остаток высушили над P2O5 (100 °C, 1÷ 2 мм.рт.ст.). Пиролиз полученной смеси солей (5,7 г) дал 3,9 г смеси, содержащей в основном олефины 17-18. Повторной перегонкой выделено 3,4 г (73%) смеси соединений 17:18 = 3:1 (ХМС, ЯМР 19F), т.кип. 83÷ 84 оС/17 мм.рт.ст.. Найдено %: 33,58; F 58,48. C11F12O2. Вычислено %: С 33,67; F 58,16. Масс-спектр: 392 [M]+ (2 изомера).
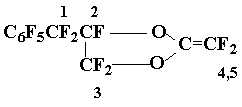
Спектр ЯМР 19F метилендиоксолана 17: AB-система с центрами 29,2 и 32,9 (2F1, JAB = 292 Гц); 49,5 (1F2); AB-система с центрами 7,4 и 10,45 (2F3, JAB = 139 Гц); AB-система с центрами 51,6 и 53,1 (1F4 + 1F5, JAB = 128 Гц); 62,1 (2o-F); 72,1 (1 p-F); 85,6 (2 m-F).
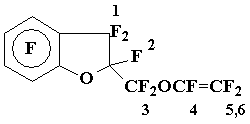
Спектр ЯМР 19F винилового эфира 18: AB-система с центрами 25,1 и 31,0 (2F1, JAB = 264 Гц); 57,3 (1F2); 9,2 (центр мультиплета, 2F3); 60,1 дд [1F4, J(F4÷транс-F5) = 112 Гц; J(F4÷F6) = 68 Гц]; 39,7 дд [1F6, J(F6÷F5) = 90 Гц]; 47,0 дд (1F5); 64,2 + 68,5 (2 o-F); 83,4 + 84,2 (2 m-F).
Список литературы
- У.Шеппард, К.Шартс, «Органическая химия фтора», «Мир», М., 1972, с.172.
- З.Н.Нудельман, «Фторкаучуки: основы, переработка, применение», М., ООО «ПИФ РИАС». Производственное издание. 2007, С. 383.
- A.P.King, C.G.Krespan, US Pat. 4384128 (1983).
- И.Л.Кнунянц, Л.С.Герман, Г.Г.Беленький, Г.И.Савичева, Пат. РФ № 519919 (1992); Б.И. 1992, N 41.
- И.П.Коленко, Т.И.Филякова, А.Я.Запевалов, Е.П.Мочалина, Л.С.Герман, В.Р.Полищук, Изв. АН СССР, Сер.хим., 1979, 667 [Bull.Acad.Sci., Div.Chem.Sci. (Eng.Trans.), 1979, 28, 622].
- а) S.Selman, US Pat. 3308107 (1967); б) US Pat. 3321517 (1967); в) US Pat. 3404162 (1968).
- D.C.England, US Pat. 3962279 (1975); C.A. 85 (1976), 160068.
- В.С.Юминов, С.В.Карцев, В.Л.Максимов, А.В.Фокин, Изв.АН СССР, Сер.хим., 1988, 392 [Bull.Acad.Sci., Div.Chem.Sci. (Eng.Trans.), 1988, 37, 311].
- С.Р.Стерлин, Э.А.Аветисян, Изв. РАН, Сер.хим., 2009, №6, с. 1292 [Bull.Acad.Sci.(Eng.Trans.), 2009, N 6, 1318].
Материал рекомендован к публикации членом редколлегии В.В. Корниловым
Fluorine Notes, 2012, 85, 5-6
