Received: October , 2012
Fluorine Notes, 2012, 85, 7-8
Role of Copper in Synthesis of Fluoroalkylsilanes by Interaction of Fluoroalkyl Halides with Silylation Agents in the Presence of a Zinc–Copper Couple
S.M. Igoumnovab, V.K. Men'shikovab, V.E. Boykoab, A.A. Tyutyunovab, S.R. Sterlina
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, V-334, GSP-1, 119991 Moscow, Russia
b"P&M-Invest", ul. Vavilova 28, 119991 Moscow, Russia
e-mail: tuytuynov@rambler.ru
Abstract: It is found that fluoroaliphatic and fluoroaromatic bromides and iodides interact with silyl carboxylates and silyl sulfonates under the conditions of the Barbier reaction in the presence of a Zn-Cu couple and form the corresponding fluoroaliphatic and fluoroaromatic silicon derivatives. Presynthesized organofluorine zinc derivatives do not interact with silylating agents in a wide temperature range irrespective of the presence or absence of copper in the reaction mixture.
Keywords: fluoroalkylsilanes, zinc-copper galvanic couple, fluoroalkylzinc halides.
Organofluorine silanes can be literally named universal reagents for introduction of organofluorine substituents of aliphatic, aromatic, or olefin series into various classes of organic and organoelement compounds [1-3]. (Trifluoromethyl)trimethylsilane (1a) (Ruppert reagent) first obtained in 1984 in the reaction of trifluoromethyl iodide and trimethylchlorosilane in the presence of tris(diethylamido)phosphine [4] attained particular popularity, as it manifests a pronounced trifluoromethylating effect under the conditions of nucleophilic catalysis [1, 5-9].
The synthetic importance of organofluorine silanes resulted in appearance of a whole number of works dedicated to development of simpler and more convenient methods of their synthesis [1]. Thus, synthesis of silane 1a was successfully completed on the basis of trifluoromethyl bromide and trimethylchlorosylane under the electrochemical reaction conditions using sacrificial zinc or aluminum anodes [10-12]. Fluoroalkylsilanes are formed at the preparative efficiency in the reaction of fluoroalkyl halides with metals (Al, Zn, Mg) in the presence of silylating agents (Me3SiCl, Me3SiOC(O)CF3, Me3SiOSO2Me) [12-17]. An alternative approach to synthesis of trifluoromethyl derivatives of silicon using ozone-safe trifluoromethane has also been described [18], but this method requires application of strong bases, e.g., potassium tert-butylate and can hardly be considered as a preparative technique for synthesis of silane 1a.
The most convenient preparative synthesis technique among the above methods of preparing organofluorine silicon derivatives is an interaction between fluoroalkyl or fluoroaryl halides and silylating agents in the presence of Zn-Cu couple obtained in situ, under the conditions of the Barbier reaction (Table 1) [15-16]. The reaction mechanism was not discussed in these works.
Table 1.
Synthesis of organofluorine silanes by the
reaction of organofluorine halides with silyl esters in the presence
of a zinc-copper couple
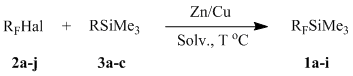
|
# |
RFHal (2a-j) |
RSiMe3 (3a-c)1 |
Solv. |
T, oC |
Yield RFSiMe3 (1a-i)2, % |
|
1 |
CF3Br (2a) |
3c |
DMF |
0 |
10 (1a) |
|
2 |
2a |
3a |
DMF |
25 |
65 (1a) |
|
3 |
2a |
3b |
DMF |
0 |
67 (1a) |
|
4 |
2a |
3b* |
NMP |
0 |
45 (1a*) |
|
5 |
C2F5I (2b) |
3a |
DMF |
20 |
41 (1b) |
|
6 |
2b |
3b |
DMF |
0 |
80 (1b) |
|
7 |
nC3F7I (2c) |
3b |
DMF |
0 |
64 (1c) |
|
8 |
nC3F7Br (2d) |
3b |
DMF |
0 |
81 (1c) |
|
9 |
iC3F7Br (2e) |
3b |
DMF |
0 |
57 (1d) |
|
10 |
C4F9I (2f) |
3b |
DMF |
0 |
60 (1e) |
|
11 |
C6F5Br (2g) |
3b |
DMF |
20 |
65 (1f) |
|
12 |
BrCF2CO2Me (2h) |
3b |
NMP |
5 |
70 (1g) |
|
13 |
BrCF2CO2Et (2i) |
3a |
DMF |
20 |
40 (1h) |
|
14 |
2i |
3b |
DMF |
0 |
38 (1h) |
|
15 |
2i |
3b |
NMP |
5 |
75 (1h) |
|
16 |
BrCF2CF2OCF=CF2 (2j) |
3b |
DMF |
0 |
50 (1i) |
1 3a: R = CF3CO2; 3b: R = CH3SO3; 3c: R = Cl. 3b* - CH3SO3SiEt3.
2 1a: RF = CF3; 1b: RF = C2F5; 1c: RF = nC3F7; 1d: RF = iC3F7; 1e: RF = C4F9; 1f: RF = C6F5; 1g: RF = CF2CO2Me; 1h: RF = CF2CO2Et; 1i: RF = CF2CF2OCF=CF2. 1a* - CF3SiEt3.
One can reasonably assume that the key intermediates in these reactions are organofluorine zinc derivatives. It is known that catalytic amounts of copper, as well as other metals with a high oxidation potential, promote formation of organozincs [19-21], including fluoroaliphatic zinc derivatives [22-23].
We found that trifluoromethyl bromide interacts with zinc (activated in situ by addition of 5 mol.% of Me3SiCl) in a DMF solution at the temperature of 60-65Ѓ‹C with formation of a mixture of (trifluoromethyl)zinc bromide (4a) and bis(trifluoromethyl)zinc (4b) (cf. the data in [24-25]). Similarly, perfluorohexylzinc bromide is obtained from perfluorohexyl bromide and zinc activated by Me3SiCl in DMF or sulfolane medium (according to 19F NMR data).
In the presence of catalytic amounts of copper (deposited in situ by addition of copper salts to a suspension of Zn in the solvent), this reaction starts already at the room temperature and occurs exothermally at an increase in the temperature to 30-32Ѓ‹C resulting in formation of a mixture of 4a and 4b as the main reaction products. However, the thus obtained mixture of trifluoromethyl zinc derivatives 4a-b does not react further with silylating agents (trimethylsilyl trifluoroacetate (3a) or trimethylsilyl mesylate (3b)) even under heating to 100Ѓ‹C irrespective of the presence or absence of copper.
One can assume on the basis of the obtained experimental data that the catalytic role of copper is determined by the very essence of a Zn-Cu galvanic couple obtained, in which copper performs a role of negative charge carrier (cf. the data in [19-21]). Reduction of RFHal by the zinc-copper couple results in formation of zinc-copper complexes 5. Their further behavior depends on the absence or presence of silyl ester 3a-b in the reaction medium: in the first case, complex 5 eliminates copper yielding organofluorine zinc derivative; in the second case, it reacts with the silylating agent forming silanes 1a-i in the following chain of transformations (Scheme 1).
Scheme 1
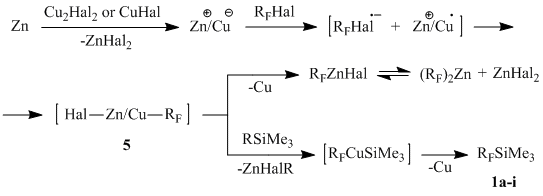
One should note that as suggested earlier in the discussion on the mechanism of direct synthesis of methylchlorosilanes MenSiCl4-n from methyl chloride and silicon in the presence of Cu2Cl2 (continuous-flow system, 300Ѓ‹C), the Si+-Cu– galvanic couple takes part in the reaction [26]. The author assumes that synthesis of methylchlorosilanes is preceded by ionization of methyl chloride on the ionized crystal lattice of the intermetallic Si-Cu alloy, which results in formation of silyl chloride and methyl copper – a source of the methyl radical. The possibility of reductive dechlorination of methyl chloride was not considered here.
Also galvanic couples formed under deposition of Ni, Cu, Ag, Au and platinum group metals on Zn, Fe or Fe2+ hydroxides are used for hydrogenation and deuteration of cinnamic acid [27] and reduction of CCl4 and chlorohydrocarbons [28 and references therein].
One can assume that the synthetic potential of galvanic couples in redox reactions goes beyond the above reactions and studies in this area appear rather promising.
Experimental
1H, 19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300 and 282 MHz, accordingly; the external standard was CDCl3 or D2O. Chemical shifts for 1H spectra are presented vs. the residual signal of the solvent (ѓВ 7.25, 4.79) and are given in ppm vs. tetramethylsilane. Chemical shifts in 19F spectra are given in ppm vs. CF3COOH. Downfield shifts are positive.
General method of synthesis of organofluorine silanes 1a-i.
Cu2Cl2 (0.5 g, 2.5 mmol) and Me3SiCl (0.5 g, 5 mmol) were successively added to a suspension of a Zn powder (6.5 g, 0.1 g-atom) in absolute DMF or NMP (see Table 1) (80 ml) and mixed for 30 min. The reaction mass was cooled to -10Ѓ‹C and silyl ester 3 (0.1 mol) was added. Then halide 2 (0.09 mol) was added under vigorous mixing and maintaining the required temperature (see Table 1). The reaction mass was mixed for 1 h and the temperature was brought to 25Ѓ‹C. The reaction product was distilled in vacuum or separated after the layering of the reaction mixture.
Spectral data of silanes are presented in the literature: 1a [18], 1b [29], 1c [29], 1d [30], 1e [30], 1f [31], 1h [17].
Trimethylsilyldifluoroacetic acid methyl ester (1g).
Boiling point: 82Ѓ‹C. Found (%): C, 39.45; H, 6.40; F, 20.57. C6H12F2O2Si. Calculated (%): C, 39.54; H, 6.64; F, 20.85. 1H NMR ѓВ: 0.55 (s, 9H, Si(CH3)3), 4.15 (s, 3H, CH3); 19F NMR ѓВ: -46.8 (s, 2F, CF2).
Trimethyl(perfluoro-2-vinyloxyethyl)silane (1i).
Boiling point: 117Ѓ‹C. Found (%): C, 31.16; H, 3.51; F, 49.07. C7H9F7OSi. Calculated (%): C, 31.11; H, 3.36; F, 49.22. 1H NMR ѓВ: 0.44 (s, 9H, Si(CH3)3); 19F NMR ѓВ: -57.8 (dd, 1F, 3JFF-trans = 112 Hz, 3JFF-cis = 68 Hz, OCF), -54.0 (s, 2F, CF2Si), -47.4 (dd, 1F, 2JFF = 90 Hz, =CF-trans), -39.8 (dd, 1F, 2JFF = 90 Hz, =CF-cis), -9.8 (s, 2F, OCF2).
Reaction of trifluorobromomethane with the zinc-copper couple.
To a Zn powder suspension (6.5 g, 0.1 g-atom) in 80 ml of anhydrous DMF 0.5 g (5 mmol) of Me3SiCl and 0.5 g (2.5 mmol) of Cu2Cl2 were successively added, the mixture was stirred for 20 min. The reaction mass was heated to 30Ѓ‹C and CF3Br was bubbled at the rate providing gas absorption. An exothermal reaction starts in 15-20 min. It is supported by increasing the rate of CF3Br bubbling so that the temperature of the reaction mass doesnЃft exceeds 60Ѓ‹C. The reaction stops in 1 h. According to 19F NMR data, the obtained solution contains 88% of the mixture of (CF3)2Zn (s, ѓВ = 35.9) and CF3ZnBr (s, ѓВ = 35.1) and 12% CF3H (d, ѓВ = -1.2; 2JFH = 79.8 Hz).
References
- G.K.S. Prakash, A.K. Yudin Chem.Rev., 1997, 97, 757-786.
- J.-A. Ma, D. Cahard Chem.Rev., 2004, 104, 6119-6146 (2008, 108, PR1-PR43).
- K. Uneyama, J.Fluor.Chem., 2008, 129, 550-576.
- I. Ruppert, K. Schlich, W. Volbach Tetrahed.Lett., 1984, 25, 2195-2198.
- R.P. Singh, J.M. Shreeve Tetrahedron, 2000, 56, 7613-7632.
- S. Roy, B.T. Gregg, G.W. Gribble, V.-D. Le, S. Roy Tetrahedron, 2011, 67, 2161-2195.
- A.D. Dilman, V.V. Levin Eur.J.Org.Chem., 2011, 831-841.
- O.A. Tomashenko, V.V. Grushin Chem.Rev., 2011, 111, 4475-4521.
- E.J. Cho, S.L. Buchwald, Org.Lett., 2011, 13, 6552-6555.
- F. Aymard, J.-Y. Nedelec, J. Perichon Tetrahed.Lett., 1994, 35, 8623-8624.
- G.K.S. Prakash, D. Deffieux, A.K. Yudin, G.A. Olah Synlett, 1994, 1057-1058.
- J. Grobe, J. Hegge ZAAC, 2008, 634, 1975-1990.
- J. Grobe, J. Hegge Synlett, 1995, 641-642.
- K.I. Petko, S.Y. Kot, L.M. Yagupolskii J.Fluor.Chem., 2008, 129, 301-306.
- S.M. Igoumnov, V.A. Vyazkov, A.V. Prokhorov. RF Patent № 2399624, 2009.
- S.M. Igoumnov, E.V. Igoumnova. Sintezy ftororganicheskikh soedinenii (Syntheses of Organofluorine Compounds), Part 1, 2nd ed., ZAO NPO P&M-Invest, Moscow, 2010.
- S. Utsumi, T. Katagiri, K. Uneyama Tetrahedron, 2012, 68, 580-583.
- G.K.S. Prakash, J. Hu, G.A. Olah J.Org.Chem., 2003, 68, 4457-4463.
- J.H. Gladstone, A. Tribe J. Chem. Soc., 1873, 26, 445-452.
- J.H. Gladstone, A. Tribe J. Chem. Soc., 1877, 31, 561-570.
- J.H. Gladstone, A. Tribe J. Chem. Soc., Trans., 1879, 35, 567-578.
- H. Blancou, P. Moreau, A. Commeyras Tetrahedron, 1977, 33, 2061-2067.
- S. Benefice-Malouet, A. Commeyras J.Fluor.Chem., 1995, 70, 103-107.
- D. Naumann, W. Tyrra, B. Kock, W. Rudolph, B. Wilkes J.Fluor.Chem., 1994, 67, 91-93.
- W. Tyrra, D. Naumann J.Prakt.Chem., 1996, 338, 283-286.
- S.S. Olenin. In: Khimiya i prakticheskoe primenenie kremniiorganicheskikh soedinenii (Chemistry and Practical Application of Organosilicon Compounds), no. 1. Leningrad, Izd. TsBTI, 1958, p. 63; A.D. Petrov, V.F. Mironov, V.A. Ponomarenko, E.A. Chernyshev. Sintez kremniiorganicheskikh monomerov (Synthesis of Organosilicon Monomers), izd. AN SSSR, Moscow, 1961, p. 39.
- C. Petrier, J-L. Luche, S. Lavaitte, C. Morat J.Org.Chem., 1989, 54, 5313-5317.
- E.J. OLoughlin, K.M. Kemner, D.R. Burris Environ.Sci.Technol., 2003, 37, 2905-2912 and lit.cit.
- R. Krishnamurti, D.R. Bellew, G.K.S. Prakash J.Org.Chem., 1991, 56, 984-989.
- V.A. Petrov Tetrahed.Lett., 2001, 42, 3267-3269.
- H.-J. Frohn, A. Lewin, V.V. Bardin J.Organomet.Chem., 1998, 570, 255-263.
Recommended for publication by Prof. S. Igoumnov
Fluorine Notes, 2012, 85, 7-8
