Received: November, 2012
Fluorine Notes, 2012, 85, 5-6
SYNTHESYS OF PERFLUOROBENZYL KETONES AND RESEARCH OF THEIR REACTIVITY
G.A.Emelianova, V.V.Berenblita, S.R.Sterlinb
aS.V.Lebedev Research Rubber Institute, 1 Gapsalskaya St., 198035 Saint-Petersburg, Russia
e-mail: emelianovgennadiy@yandex.ru
bA. N. Nesmeyanov Institute of Organoelement Compounds,Russian Academy of Sciences, 28 Vavilov St., 119991 Moscow, Russia)
Abstract:1,2-Epoxyperfluoro-3-phenylpropane was obtained by liquid phase oxidation of perfluoroallylbenzene. Under the action of SbF5 or HSO3F the epoxide was transformed into perfluoromethylbenzyl ketone and fluorosulfonyloxidifluoromethyheptafluorolbenzyl ketone respectively. The methods of preparation of perfluorovinyl ethers based on the ketones obtained as starting materials have been studied.
Keywords: 1,2-Epoxyperfluoro-3-phenylpropane, perfluoromethylbenzyl ketone, fluorosulfonyloxydifluoromethylheptafluorobenzyl ketone.
The unique chemical and thermal stability of perfluorinated rubbers of saturated row is determined by high – 107÷121 Kkal/mol – value of C-F-bond [1]. At the same time this property of organofluorine compounds doesn’t allow to cure perfluorinated rubbers by methods traditionally used for vulcanization of nonfluorinated or partially fluorinated rubbers [2].
One of the possible solutions of this problem is a preparing perfluorinated rubber with incorporated monomer bearing a functional group, which is capable to interact with nucleophilic agents. Thus the copolymer of perfluorophenylglycidyl ether with HFPO can be cured under the action of bis-phenol A di-K-salt (N2, 200 В°C/3 days) [3].
It could be expected that the introduction of fluoroalkyl substituent into aromatic ring entering the structure of perfluorinated monomers of alkyl aromatic row would increase the electrophilicity of pentafluorophenyl group, and the curing of rubbers containing monomers of such structure under the action of bifunctional nucleophiles could be achieved under more mild conditions.
In order to prepare perfluorovinyl ethers containing perfluorobenzyl group we have attempted to synthesize perfluorobenzyl ketones (4, 6).
The ketones 4 and 6 were synthesized according to the scheme comprising liquid phase oxidation of perfluoroallylbenzene (1) [4] (scheme 1)
В Scheme 1

and consecutive electrophilic isomerization of perfluoroallylbenzene oxide (2) formed under the action of SbF5 affording the ketone 4 (scheme 2) or HSO3F that led to the formation of ketone 6(scheme 3).
In the presence of catalytic amounts of SbF5 (mol. ratio 2:SbF5 = 60:1) oxide 2 forms a mixture of ketone 4 and perfluorotoluene (mol. ratio 9:1) as a result of exothermic reaction:
Scheme 2
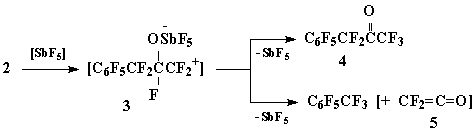
Taking into consideration the negligible amount of SbF5 taken into reaction – with respect to the quantity of perfluorotoluene obtained - the formation of toluene can be explained by the scheme of decay of intermediate carbocation 3, which comprises the elimination of stable perfluorobenzyl cation with simultaneous capture of fluoride ion from α-position and generation of difluoroketene (5).
The reaction of oxide 2 with HSO3F affording fluorosulfonyloxyketone 6 occurs under relatively mild conditions (100В°C/1.5 h) whereas the analogous reaction of HFPO demands heating up to 200Г· 220 В°C [5].
Scheme 3
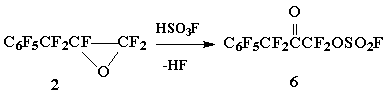
The ketone 4, alike other perfluorinated ketones, forms water soluble hydrate. In the presence of catalytic amounts of CsF at 0÷ 15°C it adds smoothly to HFPO with the formation of monoadduct (7). At the same time under more severe conditions – 80÷85°C – ketone 4 undergoes intramolecular cyclization catalyzed by CsF that leads to perfluoro-2-methylbenzofuran (8):
Scheme 4

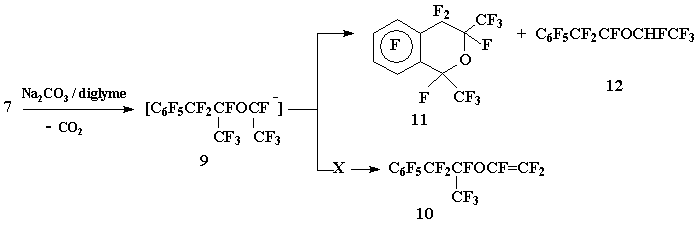
The attempt to obtain trifluorovinyl ether (10) by defluorocarbonylation of acyl fluoride 7 under the action of Na2CO3 in diglyme led to the formation of perfluoro-1,3-dimethyl-2-chromane (11) as the main reaction product as a result of nucleophilic substitution of fluorine atom in o-position of aromatic ring by carbanion (9), generated in the course of reaction, and to ОІ-(1,2,2,2-tetrafluoroethoxy)perfluoropropyl benzyne (12):
Scheme 5
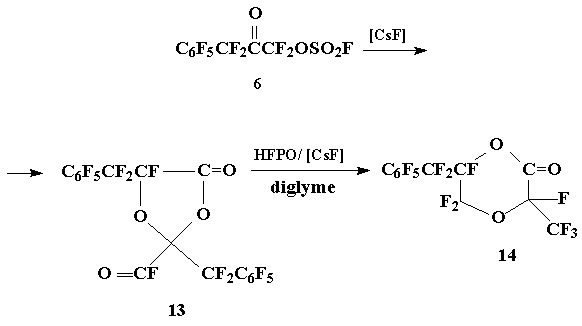
The alternative route of preparing fluorinated olefine based on oxide 2 as starting material could be a chain of transformations analogous to the scheme that was used for the synthesis of perfluoro-2-methylene-4-methyl-1,3-dioxolane [6aГ· c]. This approach comprises the conversion of ketone 6 into the dimer of О±-ketoacyl fluoride (13), the preparation of cycloadduct of (13) with HFPO, the isomerization of 2-oxo-1,4-dioxane (14) obtained in the presence of CsF into 2-fluoroformyl-1,3-dioxolane (15) and saponification of 15 with aq. Na2CO3 with subsequent pyrolysis of carboxilate formed.
4-Oxo-1,3-dioxolane 13 was obtained by degradation of fluorosulfonyloxyketone 6 under the action of CsF; the subsequent reaction of 13 with HFPO/[CsF] resulted in formation of 2-oxo-1,4-dioxane 14 (about the formation of homodimers pf perfluorinated О±-oxoacyl fluorides and their cycloadducts with HFPO see also [7Г· 9]):
Scheme 6
As it turned out the isomerization of dioxane 14 under the conditions described in [6] led to the formation of undivided mixture of the target 2-fluoroformyl-1,3-dioxolane 15 and perflyoro-2-benzofurylmethoxypropionyl fluoride (16) with the dominance of the latter (16:15 = 5:4). In this case as well as in the above examples the nucleophilic substitution of fluorine in the aromatic ring appeared to be a dominating direction of the process:
Scheme 7
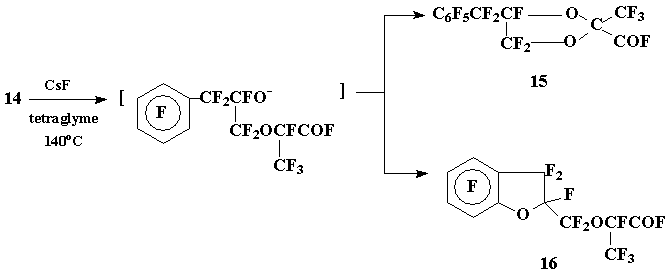
The mixture of acyl fluorides 15-16 was worked up with aq. Na2CO3; the subsequent pyrolysis of sodium carboxylates obtained afforded a mixture of trifluorovinyl ether (17) and 2-methylene-1,3-dioxolane (18), that were characterized with 19F NMR spectra:
Scheme 8

Thus we have shown that the transformations of perfluorobenzyl ketones and their derivatives that proceed with the intermediate generation of alkoxy- or cabanions are inevitably accompanied by formation of products of intramolecular nucleophilic substitution that is most probably connected with increased electrophilicity of aromatic ring in the perfluorobenzyl group structure.
Experimental
19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 282 MHz; the external standard was CDCl3 or D2O. Chemical shifts in 19F spectra are given in ppm vs. CF3COOH. Mass-spectrum were recorded VG ANALYTICAL 70-70E (70 eV) and Finnigan Polaris/GCQ Plus (70 eV) instruments.
Preparation of perfluoroallylbenzyl oxide 2
The mixture of 39 g (0,13 mol) perfluoroallylbenzene and 150 ml of Freon 113 was loaded in to 200 ml stainless steel reactor. The oxygen was fed in the reaction vessel at 140Г· 150 В°C until its consumption ceased. The rectification of the reaction mixture gave 29 g (70 %) of oxide 2, b.p. 51Г· 52 В°C/15 Torr.
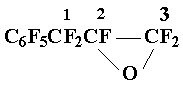
Found %: C 35,10;F 60,20. C9F10O. Calc. %: C 34,40;F 60,50.
19F NMR spectrum: AB-pattern with centra at 31.0 and 33.0 (2F3) ( JAB = 265 Hz ); 72,3 (1F2); AB-pattern with centra at 20,0 and 28,1 (2F1); 60,7 (2 o-F) 68,3 (1p-F), 81,5 (2 m-F).
Preparation of perfluoromethylbenzylketone 4
The oxide 2 (56 g, 178 mmol) was added drop wise to 0.7 g SbF5, sustaining the temperature of the reaction mixture < 40Г· 45В°C, the mixture was stirred vigorously 10 min more; according to 19F NMR data the mixture contained 90 % of ketone 4 and 10 % of perfluorotoluene. The reaction mixture was worked up with 4 ml of dry sulfolane, heated when stirred up to 105Г· 110В°C, the liquid part of the reaction mixture was decanted from tarry deposit, filtered and distilled to give 7 g of fraction (125Г· 135 В°C) that contained 4:C6F5CF3 = 1:1 and 42 g of fraction that contained 4:C6F5CF3 = 10:1; the yield of ketone 4 75 %. The analytical sample of 4 was isolated by further rectification, b.p. 141Г· 142,5В°C. Found %: F 59,94. C9F10O. Calc. %: F 60,51.
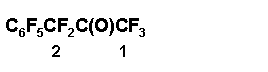
19F NMR spectrum: - 1,3 (3F1); 23,8 (2F2); 63,0 (2 o-F); 72,0 (1 p-F); 85,0 (2 m-F).
Preparation of fluorosulfonyloxydifluoromethylheptafluorobenzylketone 6
The mixture of oxide 2 (13.3 g, 42 mmol) and HSO3F (8,5 g, 85 mmol) was heated in a steel tube, protected from air moisture, at 100 В°C/1.5 h, then the reaction mixture was poured on to crashed ice, the organic layer was washed with ice water, dried with SOCl2 (the reaction mixture was kept under reflux until gas evolution ceased), the excess of SOCl2 was distilled off, the distillation of residue gave 9 g (52 %) of ketone 6, b.p. 80Г· 83 oC/7-8 Torr. Found %: C 28,10; F 47,86; S 7,73. C9F10O4S. Calc. %: C 27,41; F 48,22; S 8,12.
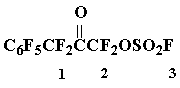
19F NMR spectrum: - 126,0 (1F3); 1,1 (2F2); 23,2 (2F1); 63,0 (2 o-F); 71,0 (1 p-F); 84,0 (2 m-F).
Preparation of acyl fluoride 7
HFPO (5 g, 30 mmol) was gradually added to a mixture of ketone 4 (9,5 g, 30 mmol), CsF (0.5 g) and 2 ml diglyme at 0В°C, stirred at 0Г· 15В°C/15 min, warmed up to ~25В°C, the low layer was separated and distilled to give 11.4 g of a fraction (52Г· 74 oC/12 Torr), that contained ~95 % acyl fluoride 7 (a mixture of diastereo isomers): the yield of 7 75 %. Further rectification gave the analytical sample, b.p. 69Г·74 oC/12 Torr (mixture of diastereomers). Found %: C 29,98; F 63,46. C12F16O2. Calc. %: C 30,00;F 63,33.
19F NMR spectrum: —102,6 (1F1); 1,0 + 2,3 (3F4); 6,3 + 6,4 (3F2); a group of signals 27,0÷ 32,0 (2F6); 48,8 + 51,5 (1F3); 61,7 (2 o-F); 65,0 (1F5); 72,0 (1 p–F); 85,5 (2 m-F).
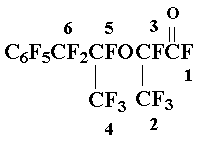
Preparation of benzofuran 8
Ketone 4 (4.5 g, 14 mmol) was added drop wise to a mixture of CsF (0.7 g) and 4 ml tetraglyme, when the exothermic reaction was over the solution obtained was stirred at 80Г· 85 oC until sedimentation ceased, the products volatile at 70 oC/1-2 Torr were evacuated into receiver (-78В°C), the condensate obtained was distilled to give 3.9 g (86 %) of benzofuran 8, b.p. 74Г· 76 oC/70 Torr. Found %: C 34,71;F 59,61.C9F10O. Calc. %: C 34,39;F 60,51.
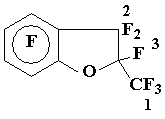
19F NMR spectrum: 5,30 + 5,35 (3F1); AB-pattern with centra 25,2 Рё 31,2, JAB = 235 Hz (2 F2); 57,0 (1F3); 64,5 + 68,7 (2 o-F); 83,6 + 84,4 (2 m-F).
The reaction of acyl fluoride 7 with Na2CO3
The acyl fluoride 7 (5.4 g, 11 mmol) was gradually added to a mixture of Na2CO3 (1.2 g, 12 mmol) Рё 15 ml of diglyme at 65Г· 70 oC, the reaction mixture was stirred at 140 В°C until gas evolution ceased, washed with dilute aq. HCl, the organic layer (4 g) was separated and distilled over H2SO4 to give 3.3 g of a mixture chromane 11 and hydride 12 (11:12 = 4:1) (ChMS).
Mass-spectrum 11 (m/z, reference): 414 [M]+ 20; 395 [M-F]+ 18; 367 [M-COF]+ 10; 345 [M-CF3]+ 100; 326 [C10F10O]+ 2; 317 [C9F11]+ 8; 295 [C9F9O]+ 2; 276 [C9F8O]+ 8; 267 [C9F7O]+ 18; 248 [C8F8]+ 15; 245 [C8F7O]+ 17; 229 [C8F7]+ 12; 226 [C8F6O]+ 8; 217 [C7F7]+ 6; 198 [C7F6]+ 30; 179 [C7F5]+ 20; 167 [C6F5]+ 2; 160 [C7F4]+ 4; 148 [C6F4]+ 5; 141 [C7F3]+ 5; 129 [C6F3]+ 3; 124 [C4F4]+ 5; 117 [C5F3]+ 5; 110 [C6F2]+ 2; 98 [C5F2]+ 2; 93 [C3F3]+ 5; 79 [C5F]+ 2; 69 [CF3]+ 31.
Mass-spectrum 12 (m/z, reference): 434 [M]+ 7; 318 [C9HF11]+ 1;В 267 [C9F7O]+ 5; 217 [C7F7]+ 100; 198 [C7F]+ 2; 167 [C6F5]+ 2; 101 [C2HF4]+ 10; 69 [CF3]+ 8; 51[CHF2]+ 3.
Preparation of 4-oxo-1,3-dioxolane 13 and 2-oxo-1,4-dioxane 14
Ketone 6 (18,3 g, 46 mmol) was added drop wise to a mixture of CsF (0.8 g) and 3 ml of diglyme and stirred until gas evolution ceased (formation of dioxolane 13 as a mixture of isomers was registered according to 19F NMR data: 27,0 + 29,0 (2FCF2); 41,2 + 44,2 (1FCF); 59,5 + 61,5 (4F o-F); 68,5 + 70,0 (2F p-F); 83,5 (4F m-F) (a signal of COF-group hadn’t been registered). The reaction mixture was loaded in to stainless steel autoclave (50 ml), the autoclave was cooled in liquid N2, evacuated, HFPO (8.3 g, 50 mmol) was condensed in to autoclave, the reaction mixture was shaken at ~25°C/48 h and distilled to give 15 g (71 %) of dioxane 14 (a mixture of isomers), b.p. 64÷ 70oC/2÷ 2,5 Torr. Found %: C 32,40; F 56,54. C12F14O3. Calc. %: C 31,44;F 58,08.

19F NMR spectrum: 24,0 + 30,0 (2F1); 46,8 + 48,0 (1F2); a group of signals 4,0Г· 16,9 (2F3 + 3F5); 35,0 + 39,2 (1F4); 61,5 + 63,0 (2 o-F); 70,8 + 72,0 (1 p-F); 85,0 (2 m-F).
The isomerization of 2-oxo-1,4-dioxane 14 under the action of CsF
The mixture of dioxane 14 (14,8 g, 32 mmol), CsF (0,4 g) and 2 ml tetraglyme was stirred at 140oC/2 h, subsequent distillation afforded a mixture of isomers 15-16 (12.0 g), b.p. 41Г· 46 oC/1,5Г· 2 Torr. Found %: C 32,11; F 57,28. C12F14O3. Calc. %: C 31,44;F 58,08.Molecular ion 458 [M]+ is present in mass-spectra of both isomers.
The preparation of vinyl ether 17 and olefine 18
The mixture of acyl fluorides 15Г· 16 was worked up with aq. Na2CO3, the aqueous solution was extracted with ether, the solvent was evaporated, the residue was dried over P2O5 (100В°C, 1Г· 2 Torr). The pyrolysis of a mixture of salts obtained (5.7 g) afforded 3.9 g of a mixture that contained mainly the olefins 17Г·18. Further distillation gave 3.4 g (73 %) of a mixture 17:18 = 3:1 (ChMS, 19F NMR), b.p. 83Г· 84oC/17 Torr. Found %: 33,58; F 58,48. C11F12O2. Calc. %: C 33,67;F 58,16.Mass-spectrum: 392 [M]+ (2 isomers).
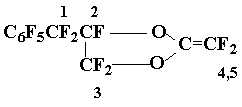
19F NMR spectrum of methylenedioxolane 17: AB-pattern with centrum 29,2 and 32,9 (2F1, JAB = 292 Hz); 49,5 (1F2); AB-pattern with centra 7,4 Рё 10,45 (2F3, JAB = 139 Hz); AB-pattern with centra 51,6 Рё 53,1 (1F4 + 1F5, JAB = 128 Hz); 62,1 (2 o-F); 72,1 (1 p-F); 85,6 (2 m-F).
19F NMR spectrum of vinyl ether 18: AB-pattern with centrums 25,1 and 31,0 (2F1, JAB = 264 Hz); 57,3 (1F2); 9,2 (centrum of multiplets, 2F3); 60,1 dd (1F4, JF4Г·trans-F5 = 112 Hz; JF4Г·F6 = 68 Hz); 39,7 dd (1F6, JF6Г·F5 = 90 Hz); 47,0 dd (1F5); 64,2 + 68,5 (2 o-F); 83,4 + 84,2 (2 m-F).
References
- W.A.Sheppard, C.M.Sharts, Organic Fluorine Chemistry (Russ.Trans.), "Mir", M., 1972, p. 17
- Z.N.Nudelman, Fluororubbers: basics, processing, application, "PIF RIAS", M., Productive Ed., 2007, p.383.
- В A.P.King, C.G.Krespan, US Pat. 4384128 (1983).
- I.L.Knunyants, L.S.German, G.G.Belenkii, G.I.Savicheva, RF Pat. 519919 (1992);
- I.P.Kolenko, T.I.Filyakova, A.Ya.Zapevalov, E.P.Mochalina, L.S.German, V.R.Polischuk, Izv. Acad. Sci. USSR, Ser. Chim., 1979, 667 [Bull.Acad.Sci., Div.Chem.Sci. (Eng.Trans.), 1979, 28, 622].
- a) S.Selman, US Pat. 3308107 (1967); b) US Pat. 3321517 (1967); c) US Pat. 3404162 (1968).
- D.C.England, US Pat. 3962279 (1975).
- V.S.Yuminov, S.V.Kartsev, V.L.Maximov, A.V.Fokin, Izv. Acad.Sci. USSR, Ser.Chim., 1988, 392, [Bull.Acad.Sci., Div.Chem.Sci. (Eng.Trans.), 1988, 37, 311].В
- S.R.Sterlin, E.A.Avetisyan, Izv. Russ. Acad. Sci., Div.Chem.Sci., 2009, Iss. 6, p. 1292 [Bull.Acad.Sci.(Eng.Trans.), 2009, N 6, 1318].
Recommended for publication by V. Kornilov
Fluorine Notes, 2012, 85, 5-6
