Received: October , 2012
Fluorine Notes, 2012, 85, 1-2
Reactions of (Perfluoroalkyl)trimethoxyborates with Electrophilic Reagents
A.A. Tyutyunovab, V.E. Boykoab, I.V. Ananyeva, V.A. Karnoukhovaa, K.A. Lyssenkoa, S.M. Igoumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, V-334, GSP-1, 119991 Moscow, Russia
b"P&M-Invest", ul. Vavilova 28, 119991 Moscow, Russia
e-mail: tuytuynov@rambler.ru
Abstract: It is shown that interaction of (perfluoroalkyl)trimethoxyborates with the overall formula of K[RFB(OMe)3] (RF = CF3, C2F5) with acyl chlorides, perfluoroacyl fluorides, etherate of BF3, and sulfuric acid results in formation, as dependent on the nature of the electrophile, of (perfluoroalkyl)dimethoxyboranes, (perfluoroalkyl)trifluoroborates, (perfluoroalkyl)boroxines, and mixed anhydrides of perfluoroalkylboronic and sulfuric acids (boroxinesulfates).
Keywords: perfluoroalkylborates, perfluoroalkylboranes, trifluoroborates, boroxinesulfate, boroxines, boronic acids.
The ever growing interest towards perfluoroalkyl compounds of boron is due to the ever expanding sphere of their practical application, particularly, as fluoroalkylating agents, electrolytes, ionic liquids, weakly coordinating anions, and ligands [1].
At the same time, despite the availability of such fluoroaliphatic derivatives of boron as alkali fluoroalkyltrimethoxyborates, the data on their reactivity are rather fragmentary. Thus, interaction with electrophilic reagents is represented only by demethoxylation under the action of silylating (ClSiMe3), sulfonylating (MeSO2Cl), and methylating (TsOMe, TfOMe) agents with formation of (fluoroalkyl)dimethoxyboranes as the only reaction products [1–2].
Reactions of borates with the overall formula of K[RFB(OMe)3] (1a-b) [RF = CF3 (1a); RF = C2F5 (1b)] with electrophiles of different nature including Lewis and Broensted acids were studied for further research of the chemical behavior of fluoroalkyltrialkoxyborates towards electrophilic reagents.
Reactions of potassium (perfluoroalkyl)trimethoxyborates 1a-b with acyl halides
Salts 1a-b undergo demethoxylation in a reaction with acetyl chloride, benzoyl chloride, or benzoyl bromide and form fluoroalkyldimethoxyboranes (cf. with the data of [2]). It should be noted that as opposed to AcCl and BzBr, easily reacting at ~ 25°C, interaction between borates 1a-b and BzCl requires heating up to 55-60 °C.
Scheme 1
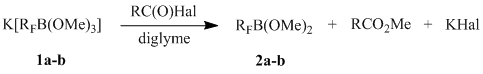
|
Nos. |
RF 1a-b |
RC(O)Hal |
Temp., oC |
Time, h |
Yield 2a-b, % |
|
1 |
CF3 (1a) |
AcCl |
25 |
3 |
80 (2a) |
|
2 |
CF3 (1a) |
BzBr |
25 |
3 |
80 (2a) |
|
3 |
CF3 (1a) |
BzCl |
25 |
4 |
10 (2a) |
|
4 |
CF3 (1a) |
BzCl |
60 |
1.5 |
70 (2a) |
|
5 |
C2F5 (1b) |
BzCl |
60 |
1.5 |
70 (2b) |
The composition of reaction products changes profoundly when perfluoroalkanoyl fluoride is used as the acylating agent. In this case, the reaction products are mono- and difluoroborates 3 and 4 at the ratio of 2 : 1 (according to the data of 19F NMR).
Scheme 2
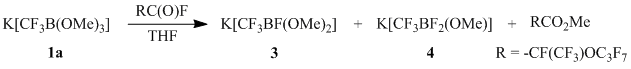
The ratio of salts 3 and 4 does not change when the reaction occurs in the temperature range of –75÷25 °C. Obviously, formation of fluoroborates 3 and 4 occurs as a result of interaction of the initially formed (trifluoromethyl)dimethoxyborane (2a) with KF, also being a reaction product, which is confirmed by countersynthesis of salts 3–4 from borane 2a and KF.
Scheme 3
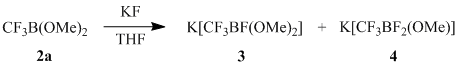
An increase in the amount of perfluoroacylfluoride introduced into the reaction results in the further substitution of methoxy groups by fluoride up to formation of (perfluoroalkyl)trifluoroborates 5a-b as the only reaction products when three equivalents of perfluoroacyl fluoride are used.
Scheme 4

Reactions of potassium (perfluoroalkyl)trimethoxyborates 1a-b with boron trifluoride etherate
We showed that the reaction of BF3•OEt2 with borates 1a–b is a convenient preparative method of trifluoroborates synthesis 5a-b, though the yield of salts 5 in this case is somewhat lower as compared to the reaction in scheme 4.
Scheme 5
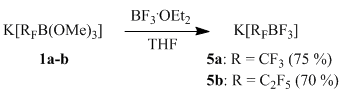
As opposed to the above method of synthesis of trifluoroborates 5a–b in the reaction of hydrofluoric acid and salts 1a–b [3–4], the suggested method is more convenient, as it allows carrying the reaction out in glass equipment. According to the data of [5], BF3 was used to obtain sodium hydrotrifluoroborate, but application of BF3 in synthesis of organotrifluoroborates has not been described earlier [6].
Reactions of potassium (perfluoroalkyl)trimethoxyborates 1a-b with sulfuric acid
It is known that the reaction between mineral acids (HCl, H2SO4, etc.) with borate salts containing various organic groups leads to formation of the corresponding boronic acids that interact with dehydrating reagents, e.g., concentrated sulfuric acid, and also in a number of cases, under drying or in the course of crystallization, forming the corresponding boroxines [7]. In particular, this method was used to obtain fluoroaromatic boronic acids and the corresponding boroxines [8–9]. However, perfluoroalkylboronic acids and their boroxines are not as yet described in the literature [1].
We found that interaction of potassium (trifluoromethyl)trimethoxyborate (1a) with concentrated sulfuric acid in a diglyme solution in a high yield results in formation of (trifluoromethyl)boroxinesulfate 6 isolated from the reaction mixture in the form of a solvate containing two molecules of water and two molecules of diglyme.
Scheme 6
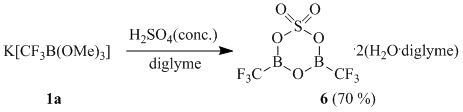
Formation of boroxinesulfates in such reactions has not been registered earlier. The structure of solvate complex 6 is confirmed by the data of elemental analysis and X-ray diffraction study (Fig. 1).
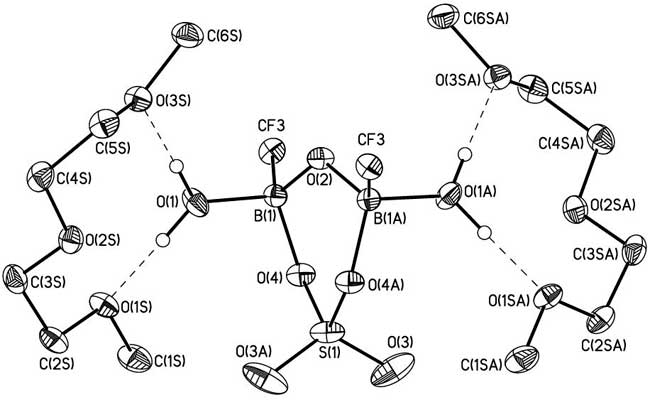
Fig. 1. The general view of 6 in representation of non-hydrogen atoms by probability ellipsoids of thermal vibrations. Hydrogen atoms of diglyme molecules are omitted for clarity.
At the same time, potassium (pentafluoroethyl)trimethoxyborate (1b) forms under similar conditions (pentafluoroethyl)boroxine 7b isolated in the form of a solvate that also contains two molecules of water and two molecules of diglyme according to the data of X-ray diffraction study (Fig. 2) and elemental analysis.
Scheme 7
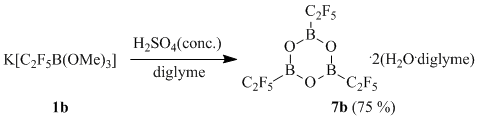
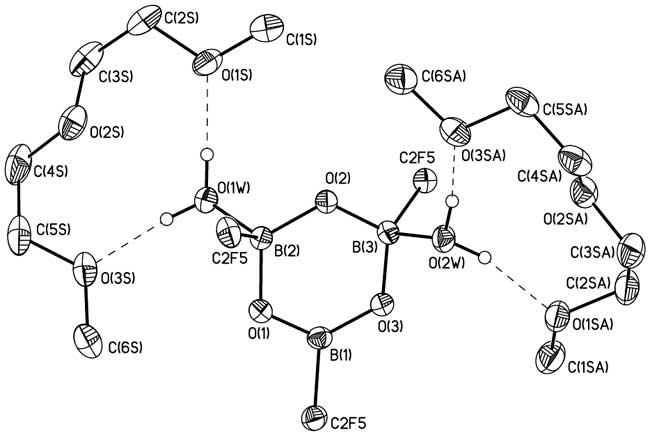
Fig. 2. The general view of 7b in representation of non-hydrogen atoms by probability ellipsoids of thermal vibrations. Hydrogen atoms of diglyme molecules are omitted for clarity.
The composition of the products of a reaction between 1a–b and sulfuric acid changes in the case of a reaction under reduced pressure (10 torr). In this case, irrespective of borate salt 1a-b used in the reaction, (perfluoroalkyl)boroxines 7a-b and (perfluoroalkyl)boranes 8a-b are formed.
Scheme 8
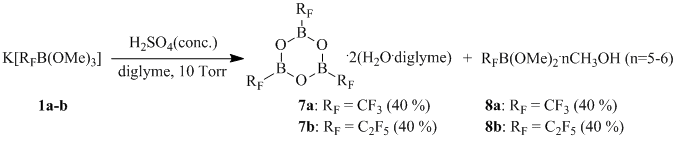
One should point out that the reaction between sulfuric acid and salt 1b under reduced pressure (10 torr) in the absence of a solvent leads to formation of pentafluoroethylboronic acid distilled from the reaction mixture in the form of a methanol adduct 9.
Scheme 9
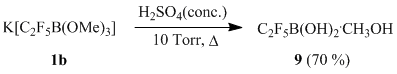
Compound 9 represents a colorless sirupy liquid and can be considered as an ansolvate acid H[C2F5B(OH)2(OMe)] [10].
It is of interest that dissolution of compound 9 in diglyme in the absence of any dehydrating reagents results in practically immediate formation of (pentafluoroethyl)boroxine 7b that is precipitates in the form of crystals.
Scheme 10
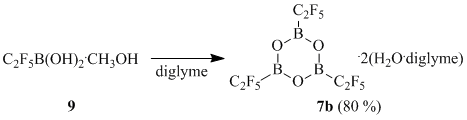
A similar reaction of borate 1a resulted in formation of a negligible amount of a compound with an unclear structure containing, according to the data of 19F NMR, a CF3BF2 fragment, which in our opinion, is an indirect proof of degradation of the trifluoromethyl group.
Reactions of potassium (perfluoroalkyl)trimethoxyborates 1a-b with water
(Perfluoroalkyl)trimethoxyborate salts 1a-b in water undergo deboronation with formation of the corresponding hydroperfluoroalkanes, which was earlier observed by other researchers [11]. Therefore, their spectral data obtained in a D2O solution [2, 12] are most probably erroneous and characterize the hydrolysis product. Thus, we showed that the main products of an addition reaction between water or deuterated water and potassium (perfluoroalkyl)trimethoxyborates 1a-b in a THF solution are hydroperfluoroalkanes (or deuteroperfluoroalkanes); the relative content of perfluoroalkylborates 10a-b (characterized using 19F NMR) does not exceed 10 %.
Scheme 11
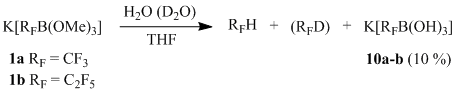
At the same time, one should point out that no deboronation is observed for a prolonged time (for about a month) when (trifluoromethyl)boroxinesulfate 6, (perfluoroalkyl)boroxines 7a-b, and ansolvate acid 9 are dissolved in water or an aqueous–ether mixture. Aqueous solutions of these compounds are characterized by high acidity and are titrated by aqueous alkali solutions.
|
Compound |
pH of aqueous solution |
|
6 |
2.8 |
|
7a |
2.5 |
|
7b |
3.2 |
|
9 |
2.7 |
Thus, we showed that the fluoroalkyl group remains at the boron atom in the interaction of borates 1a-b with acyl chlorides, perfluoroacyl fluorides, etherate of BF3, and sulfuric acid and (perfluoroalkyl)dimethoxyboranes, (perfluoroalkyl)trifluoroborates, (perfluoroalkyl)boroxines, and mixed anhydrides of perfluoroalkylboronic and sulfuric acids (boroxinesulfates) are formed, as dependent on the nature of the electrophile.
Experimental
19F, and 11B NMR spectra were recorded using a Bruker AVANCE-300 and 400 spectrometer at 282, and 128 MHz, accordingly, in D2O or with D2O as an external standard. Chemical shifts in 19F spectra are given in ppm vs. CFCl3. Chemical shifts in 11B spectra are given in ppm vs. BF3.OEt2.
Potassium (perfluoroalkyl)trimethoxyborates 1a-b were obtained earlier according to the earlier described method using THF or diglyme as a solvent [2, 4]. 93-95 % sulfuric acid was used in reactions.
Synthesis of (perfluoroalkyl)dimethoxyboranes 2a-b.
Benzoyl chloride (7.0 g, 50 mmol) was added dropwise to a solution of salt 1a or 1b (50 mmol) in absolute diglyme (50 ml) under mixing at the temperature of 55-60°C under vacuum (10 torr) and the volatile reaction product was distilled into a receiver (-78°C). After all benzoyl chloride was added, the reaction mixture was mixed for 1.5 h at 55-60°C and then the pressure was adjusted to the atmospheric pressure by adding an inert gas.
(Trifluoromethyl)dimethoxyborane (2a). Yield, 70 %.
(Pentafluoroethyl)dimethoxyborane (2b). Yield, 70 %.
Synthesis of potassium (perfluoroalkyl)trifluoroborates 5a-b.
A: Perfluoro-2-propoxypropionyl fluoride (49.8 g, 150 mmol) was added to a solution of salt 1a or 1b (50 mmol) in absolute THF (50 ml) under cooling by cold water. The reaction mixture was mixed for 3 h at 50°C and evaporated to dryness under vacuum (10 torr). The obtained solid deposition was recrystallized.
Potassium (trifluoromethyl)trifluoroborate (5a). Recrystallized from isopropanol. Yield, 95 %.
Potassium (pentafluoroethyl)trifluoroborate (5b). Recrystallized from a heptane–isopropanol mixture. Yield, 93 %.
B: BF3•OEt2 (7.1 g, 50 mmol) was added dropwise to a solution of salt 1a or 1b (50 mmol) in absolute THF (50 ml) under mixing at the temperature of -30°C. The reaction mixture was heated to the room temperature ~25°C for 30 min and mixed at the room temperature for 3 h, evaporated under vacuum (10 torr) and the residue was recrystallized. The yield of 5a, 75 %; that of 5b, 70 %.
Interaction of salts 1a-b with H2SO4 in a diglyme solution.
H2SO4 (5.3 g, 50 mmol) was added dropwise to a solution of salt 1a or 1b (50 mmol) in absolute diglyme (50 ml) under mixing at the temperature of -50°C. The reaction mixture was heated to the room temperature ~25°C and mixed for 30 min. Water (5 ml) was added and the whole was mixed for another 30 min. The deposit was filtered, the solvent was evaporated under vacuum (0.5 torr) at the temperature not above 25-30°C, the residue was dissolved in chloroform (50 ml). The obtained solution was filtered, evaporated in air (in a glass). The crystals formed were filtered and dried under vacuum (0.5 torr).
A complex of (trifluoromethyl)boroxinesulfate with diglyme and water (6). Yield, 70 %. 19F NMR δ: -76.5 (br. s., CF3); 11B NMR δ: -0.7 (br. s). Found (%): C, 29.00; H, 5.58; B, 3.75; F, 19.79; S, 5.57. C14H32B2F6O13S. Calculated (%): C, 29.19; H, 5.60; B, 3.75; F, 19.79; S, 5.57.
A complex of (pentafluoroethyl)boroxine with diglyme and water (7b). Yield, 75 %. 19F NMR δ: -83.4 (s, 3F, CF3), -135.4 (s, 2F, CF2); 11B NMR δ: 3.3 (br. s). Found (%): C, 29.19; H, 4.50; B, 4.37; F, 38.37. C18H32B3F15O11. Calculated (%): C, 29.14; H, 4.35; B, 4.37; F, 38.41.
Interaction of salts 1a-b with H2SO4 in a diglyme solution under vacuum.
H2SO4 (5.3 g, 50 mmol) was added dropwise to a solution of salt 1a or 1b (50 mmol) in absolute diglyme (50 ml) under mixing at the temperature of ~25°C under vacuum (10 torr) and the volatile reaction products were distilled into a receiver (-78°C). After all H2SO4 was added, the reaction mixture was mixed for 30 min and then the pressure was adjusted to the atmospheric pressure by adding an inert gas.
Water (5 ml) was added to the reaction mixture, the whole was mixed for 30 min, the deposit was filtered, the solvent was evaporated under vacuum (0.5 torr) at the temperature not above 25–30°C and the residue was dissolved in chloroform (50 ml). The obtained solution was filtered and left to evaporate slowly in air (in a glass). The crystals formed were filtered and dried under vacuum (0.5 torr).
The distillate obtained in the experiment was distilled, Tboil = 55-70°C. Diglyme remained in the still liquor (about 15 wt. %).
A complex of (trifluoromethyl)boroxine with diglyme and water (7a). Yield, 40 %. 19F NMR δ: -76.8 (br. s., CF3); 11B NMR δ: -0.3 (br. s). Found (%): C, 30.32; H, 5.45; B, 5.57; F, 28.78. C15H32B3F9O11. Calculated (%): C, 30.44; H, 5.45; B, 5.48; F, 28.89.
Interaction between salt 1b and H2SO4 under vacuum.
H2SO4 (40 g, 376 mmol) was added dropwise to dry salt 1b (13.1 g, 50 mmol) under mixing and cooling to the temperature of 5-10°C under vacuum (10 torr) taking care that the salt is not pulverized. Further, the mixture is heated to the temperature of 100-150°C, in the course of which a colorless viscous liquid distilled in the range of 60-80°C is distilled.
Pentafluoroethylboronic acid, a complex with methanol (9). Yield, 70 %. 19F NMR δ: -84.9 (s, 3F, CF3), -136.3 (s, 2F, CF2). Found (%): C, 18.93; H, 3.24; B, 5.32; F, 48.38. C3H6BF5O3. Calculated (%): C, 18.39; H, 3.09; B, 5.52; F, 48.49.
Crystallographic data: CCDC 889080 and 889081 contain the supplementary crystallographic data for 6 and 7b. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge, CB21EZ, UK; or deposit@ccdc.cam.ac.uk).
References
- N.Yu. Adonin, V.V. Bardin Russ. Chem. Rev., 2010, 79, 757-785.
- A.A. Kolomeitsev, A.A. Kadyrov, J. Szczepkowska-Sztolcman, M. Milewska, H. Koroniak, G. Bissky, J.A. Barten, G.-V. Roschenthaler Tetrahed. Lett., 2003, 44, 8273-8277.
- H.-J. Frohn, V.V. Bardin ZAAC, 2001, 627, 15-16.
- G.A. Molander, B.P. Hoag Organomet., 2003, 22, 3313-3315.
- H.I. Schlesinger, H.C. Brown, J.R. Gilbreath, J.J. Katz JACS, 1953, 75, 195-199.
- S. Darses, J.-P. Genet Chem. Rev., 2008, 108, 288-325.
- D.G. Hall. Boronic Acids. Preparation and Applications in Organic Synthesis and Medicine. WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. 2005.
- H.-J. Frohn, N.Y. Adonin, V.V. Bardin, V.F. Starichenko ZAAC, 2002, 628, 2827-2833.
- N.Yu. Adonin, V.V. Bardin, U. Florke, H.-J. Frohn ZAAC, 2005, 631, 2638-2646.
- H. Meerwein Jus. Lieb. Ann. Chem., 1927, 455, 227-253.
- N.Yu. Adonin, V.V. Bardin, H.-J. Frohn ZAAC, 2007, 633, 647-652.
- T. Knauber, F. Arikan, G.-V. Roschenthaler, L.J. Gooben Chem. - A Eur. J., 2011, 17, 2689-2697.
Recommended for publication by Prof. S. Igoumnov
Fluorine Notes, 2012, 85, 1-2
