Received: June, 2011
Fluorine Notes, 2012, 84, 1-2
POLYFLUORINATED CYCLOHEXADIENONES: SUITABLE INTERMEDIATES FOR THE SELECTIVE SYNTHESIS OF COMPLEX ORGANOFLUORIC SUBSTANCES
L.S.Kobrina, V.N.Kovtonyuk
N.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry of the Siberian Branch of Russian Academy
of Sciences, Novosibirsk, Russia
E-mail: kovtonuk@nioch.nsc.ru
Abstract: The article summarizes the results of studies on the reactivity of polyfluorinated cyclohexadienones: nucleophilic reactions, cycloadditions, and photochemical transformations. The use of polyfluorinated cyclohexadienones for synthons in the synthesis of a wide range of earlier inaccessible fluorinated organic substances (derivatives of polyphenolic esters, arylacetic acids, naphthalene, and anthraquinone) containing carboxylic groups, cyclohexene carboxylic acids, and some fluorinated heterocyclic substances are discussed.
Keywords: Organofluoric substances, cyclohexadienones, nucleophilic reactions, cycloaddition, polyphenyl esters, arylacetic acid.
The functionalization of fluorinated compounds is of fundamental importance both because of the limited possibility for the synthesis of such derivatives by direct introduction of fluorine atoms into organic compounds containing functional groups, and because of the difficulties related with the introduction of functional groups into fluorinated compounds, that requires multistage synthesis process [1]. We disclose a novel promising approach to the synthesis of multifunctional organofluoric substances through the conversion of some comparatively easy accessible polyfluoroaromatic compounds to highly reactive and easily modifiable polyfluorinated cyclohexadienones [2].
The great interest in polyfluorinated cyclohexadienones is because of their high and diverse reactivity due to the presence of numerous reactive centres in those molecules: mobile fluorine atoms linked to their double bonds, 1,3-diene systems in 2,4-cyclohexadienones, carbonyl groups, and reactive substituents at sp3-hybridized carbon atoms.
Nucleophilic or photochemical reactions and cycloaddition were shown to be the most effective methods for the modification of polyfluorinated cyclohexadienones.
Synthesis of polyfluorinated cyclohexadienones
Polyfluorinated cyclohexadienones with various substituents in their geminal units are producible with good yields from polyfluorinated phenols, naphthols or their salts by halogenation [3] or oxidation [4-10]. (Figure 1)

Figure 1
For the manufacture of polyfluorinated cyclohexadienones with phenyl groups at their saturated carbon atoms a new convenient method was proposed that was based on the selective oxidative ortho-arylation of perfluorinated phenols or naphthols [11], and involved the interaction of appropriate alkali metal phenolates or naphtholates with PhPb(OAc)3.
In the case of sodium pentafluorophenolate the reaction does not stop at the stage of cyclohexadienone 1 formation, and nucleophilic-mobile fluorines at position 3 are substituted by pentafluorophenoxy or hydroxy groups resulting eventually in cyclohexadienone 2 or 3 [11] (Figure 2).

Figure 2
The reactions between sodium or potassium heptafluoro-1-naphtholate and ArPb (OAc)3 or Ph2Pb (OAc)2 result in corresponding 2-aryl-1-oxoheptafluoro-1,2-dihydronaphthalene 4. (Figure 3)
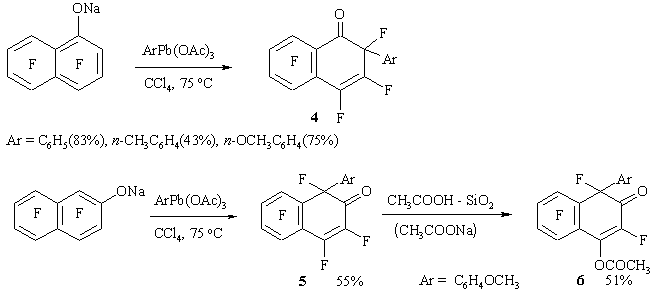
Figure 3
Sodium heptafluoro-2-naphtholate reacts with lead p-anisyltriacetate to form a mixture with predominant content of substance 5; and its chromatography on silica gel deactivated by acetic acid, results in 2-oxodihydronaphtalene 6 with yield 51%.
Nucleophilic substitution and reduction
The reactions between nucleophilic reagents and cyclohexadienones, that contain halogens at their saturated carbon atoms only, result, as a rule, in their reduction to phenol derivatives or in halogen substitution, and formation of corresponding substituted cyclohexadienones [12,13].
In contrast, polyfluorinated cyclohexadienones react easily with nucleophiles and one or two fluorines at their double bond are then replaced as a consequence both of very В high reactivity in nucleophilic reactions of fluorinated compounds with double bonds, and В as well of the double bond activation due to its conjugation with carbonyl groups [14].
Due to the carbonyl group effect, the fluorine at position 3 of polyfluorinated 2,4-cyclohexadienone is substituted much easier than that at position 5, and this result is consistent with MO calculations of charge distribution in polyfluorinated 2,4-cyclohexadienone [15]. Such reactions, usually with high yields, are exemplified in Figure 4 [16-19]. (Figure 4)
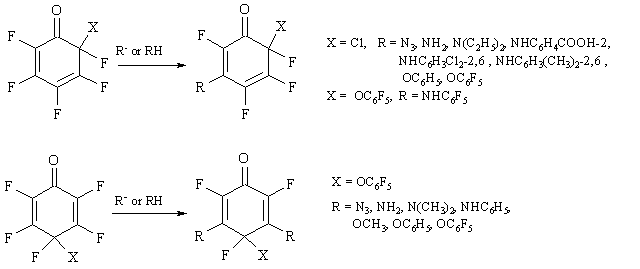
Figure 4
It should be noted that pentafluorophenoxyl group at position 3 of polyfluorinated 2,4-cyclohexadienonea 2 is easily substituted for a nucleophile at mild conditions. For example, cyclohexadienone 2 boiling with methanol results in the formation of the corresponding methoxy derivative 7 [19]. (Figure 5)

Figure 5
It is common for cyclohexadienones containing halogen at their saturated carbon atoms that being reduced they convert to phenols. When two different halogens, e.g. fluorine and chlorine, are bound to a saturated carbon atom, chlorine is always the first to be split-off under reduction. The use of this method in the synthesis of polyfluorinated phenols is the most advantageous when combined with the initial nucleophilic substitution of fluorine atom at the double bond of polyfluorinated cyclohexadienone as illustrated by examples [11,16,17,20] in Figure 6.
The method proposed for the manufacture of fluorinated phenols by the above mentioned sequence of reactions, is an example of a general strategy for the synthesis of polyfunctional aromatic compounds. At the first stage polyfluorinated phenols are converted to corresponding cyclohexadienones, which are easily and selectively modified by nucleophilic substitution of fluorines, and the cyclohexadienones thus modified are then reduced to polyfluorinated phenols containing various substituents.

Figure 6
Nucleophilic reactions between cyclohexadienone 8 and phenols [21] or tetrafluororezorcin [22] were used in the synthesis of some polyfluorinated polyphenyl esters with nitro-, amino-, or carboxyl groups, branched or linear polyfluorinated polyphenyl esters with five or eight carbocyclic fragments in their molecules.
Cyclohexadienone 8 reacts with two equivalents of sodium pentafluorophenolate at room temperature with the formation of 3,5-substituted cyclohexadienone 9. The latter undergoes isomerisation at 50-70В°C resulting in dienone 10a. Using four equivalents of phenol in the presence of potassium carbonate at 70В°C, one can produce cyclohexadienone 11a-f with phenoxy groups both in aromatic and dienone parts of the molecule (Figure 7). The rearrangement of cyclohexadienone 9 probably follows intramolecular mechanism. An indirect argument in favour of such mechanism is the absence of reaction products with the ortho-quinoid structures (quite stable under those conditions [23]), which were to be formed, would the mechanism of isomerisation involve dissociation-recombination of phenoxyl radicals.
Cyclohexadienone 11a-f can be reduced to 3,5-diaryloxy phenols 12a-f that form polyfluorinated polyphenyl esters 13a-c in reactions with hexafluorobenzene, or 13d-f in the reactions with octofluorotoluene [22].
Two equivalents of 2,3,4,5,6-pentafluoro-6-chloro-2 ,4-cyclohexa-dienone 14 react with tetrafluororezorcin at room temperature resulting in 1,3-bis(4-chlorotetrafluoro-3-oxo-1,5-cyclohexadienyloxy)-tetrafluorobenzene 15 with the yield 90%.
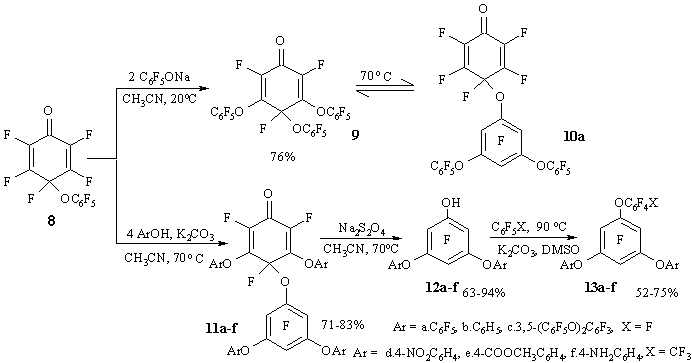
Figure 7
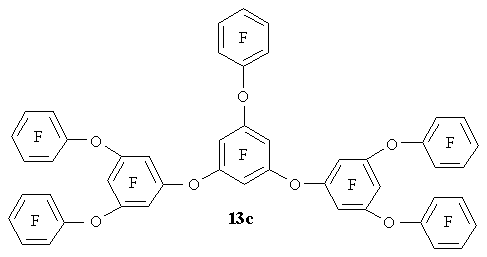
The reduction of 15 with sodium dithionite results in tetrafluoro-1,3-bis-(tetrafluoro-3-hydroxyphenoxy)benzene 16 [21], the former being also produced by the reaction of cyclohexadienone 8 with tetrafluororezorcin. The step-by-step reduction of 15 to 16, the formation of disodium salt of the latter, and its reaction with polyfluorinated octafluorotoluene result in polyfluorinated polyphenyl ether 17. (Figure 8)

Figure 8
The reaction of a nucleophile, such as triphenylphosphine, withВ polyfluorocyclohexadienone 2 leads to an interesting and unexpected result [24]. The main product of this reaction is cyclohexenedione 19, with structure that according to X-ray diffraction (XRD), contains CF2 and two carbonyl groups. Besides, in this compound phenyl group and pentafluorophenoxyl are in neighbouring positions, while in the original cyclohexadienone 2, they occupy positions 6 and 3, respectively. (Figure 9)

Figure 9
Figure 10 illustrates the likely sequence of reactions leading to the formation of compound 19.
At the first step triphenylphosphine replaces pentafluorophenoxyl group at position 3 of the original cyclohexadienone 2. After that pentafluorophenoxyl anion replaces fluorine at position 5 of the new cyclohexadienone with the formation of phosphonium fluoride A, that В undergoes isomerisation (via the formation of oxirane) to phosphonium salt B, further converted to zwitterion 18 with two CF2 groups. This compound appears to be unstable, it adds water and splits-off HF resulting in stable dione 19. (Figure 10)
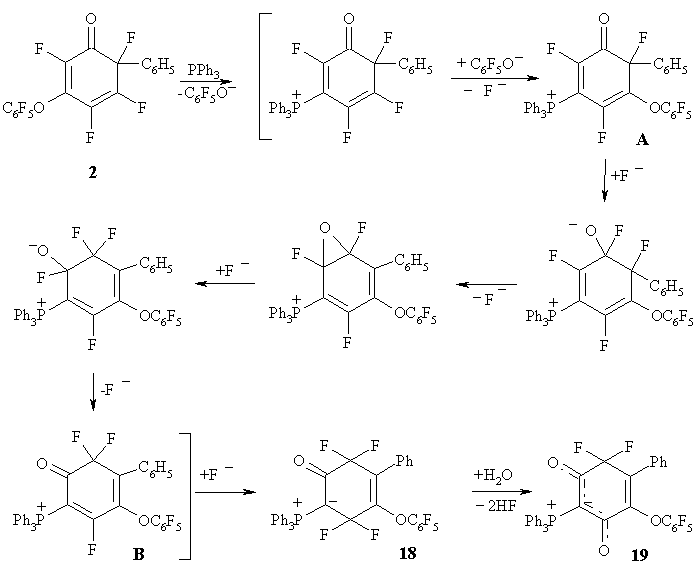
Figure 10
Reactions involving carbonyl groups
Reactions with phenylhydrazines
It is well-known that the reactions of non-fluorinated cyclohexadienone with nucleophiles occur with the participation of double bonds and carbonyl groups [25].
The transformations of cyclohexadienones by their carbonyl groups are represented, best of all, by their reactions with hydrazine derivatives [26], or with organometallic compounds [25.27].
The study on reactions between polyfluorinated cyclohexadienones and hydrazine derivatives, or organometallic compounds make it possible to evaluate the relative reactivity of two electrophilic centres available in those compounds, i.e. fluorinated double bond and carbonyl group.
Cyclohexadienone 8 in acetonitrile reacts with phenyl- or pentafluorophenylhydrazine resulting in cyclohexadienone 20 that is a product of nucleophilic substitution of fluorine atom at the double bond in the original dienone 8, then it disproportionates resulting in a mixture of corresponding 3-arylazotetrafluoro-phenols 21a, b and pentafluorophenol at equal ratio. The carbonyl group of polyfluorinated cyclohexadienone obviously does not participate in this reaction. (Figure 11)
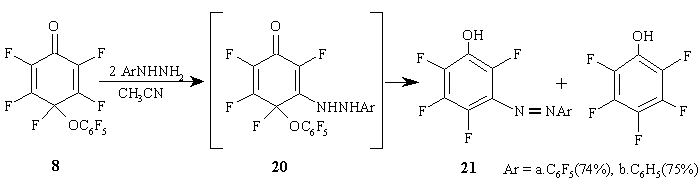
Figure 11
Since the acids are known to catalyze the reactions of arylhydrazines by carbonyl groups of cyclohexadienones [28, 29], a study was made on the interaction of polyfluorinated cyclohexadienones with complexes of phenyl- or pentafluorophenylhydrazine and aluminium chloride.
Cyclohexadienone 8 reacts with those reagents only with the participation of the carbonyl group to give the corresponding azobenzenes 22-24 [30]. During the azobenzene formation either fluorine or pentafluorophenoxyl group perform as groups leaving the cyclohexadienone geminate node. In the reactions with aluminium chloride/pentafluorophenylhydrazine complex the main reaction product is formed by the elimination of fluorine atom, while with the complex of phenylhydrazine both reaction paths are realized at nearly equal proportions. (Figure 12)
Such difference in the behaviour between phenyl- and pentafluorophenylhydrazine is probably due to the interaction of aromatic dienone substituent and hydrazine aromatic ring in the transition state. It is very likely [30], that initially formed complexes dienone/arylhydrazine/aluminium chloride have "sandwich" structure where arylhydrazine is possibly placed either on the side of aromatic fragment located in the cyclohexadienone geminate node A, or on its opposite side B. Those A and B complexes form relevant intermediates of type C and D. In the first case C pentafluorophenoxyl group will be leaving, and in the second D fluorine atom will be leaving [30].
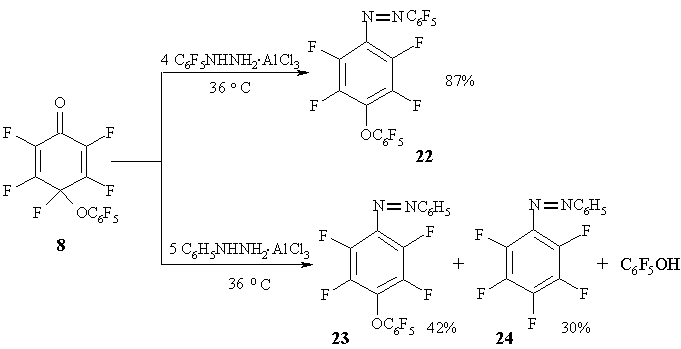
Figure 12
Intermediates with non-fluorinated phenylhydrazine type C can be further stabilized by p-interaction between the fluorinated and non-fluorinated aromatic rings (formation of p-complexes between fluorinated and nonfluorinated aromatic compounds is well known [31]), resulting, ultimately, in the splitting off pentafluorophenoxyl group from the dienone geminate node. In the case of fluoroarylhydrazine the interaction between hydrazine and dienone fluorinated aromatic rings results in the destabilization of complex C and formation of transient state D, В more favourable for the fluorine atom split-off. (Figure 13)
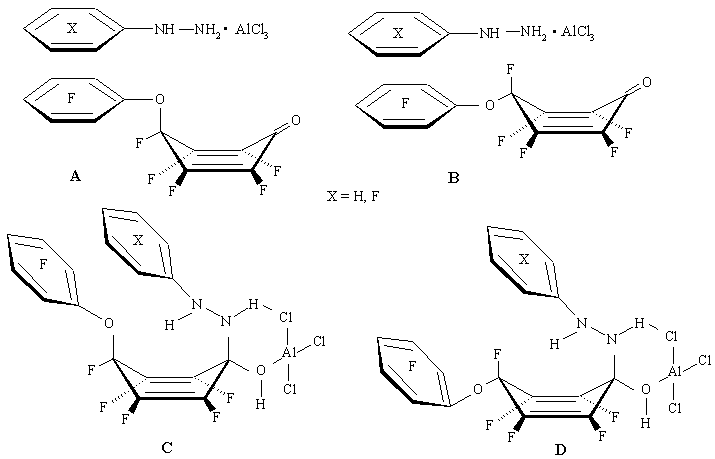
Figure 13
Arylhydrazine/aluminium chloride complexes can be used in the synthesis of various fluorinated azocompounds 25-27, as shown in Figure 14.
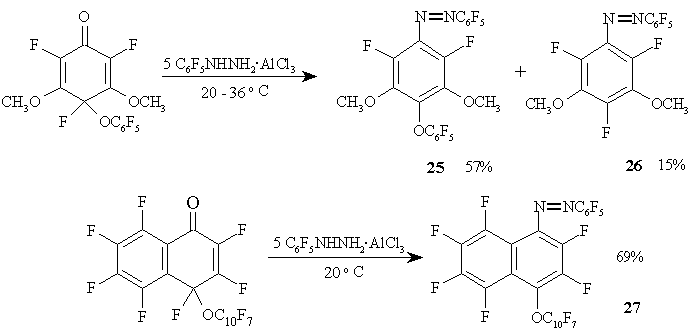
Figure 14
Reactions with organometallic compounds
The reactions between cyclohexadienone 8 and n-butyl lithium or n-butyl magnesium-bromide also occur by the carbonyl group resulting in 1-butyl-4-pentafluoro-phenoxy-2,3,4,5,6-pentafluoro-cyclohexa-2,5-diene-1-ol 28 [30]. (Figure 15)
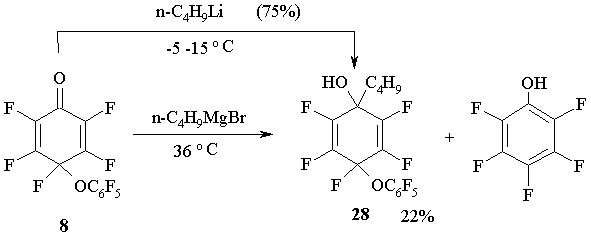
Figure 15
Therefore, polyfluorinated 2,5-cyclohexadienones react with nucleophiles exclusively by the carbonyl group, despite the presence of nucleophilic-mobile fluorine atoms at the double bond of polyfluorinated cyclohexadienone.
Photochemical transformations
Unlike the photolysis of non-fluorinated phenylcyclohexadienones, that occurs with transformations not involving carbonyl groups [32-35], photochemical reactions of polyfluorinated phenylcyclohexadienones in hexane solution proceed with the participation of both carbonyl group and phenyl fragment located in the geminate node, they result in the formation of polyfluorinated furan derivatives: dibenzofuran 29 and benzonaphthofuran 30 [36]. (Figure 16)
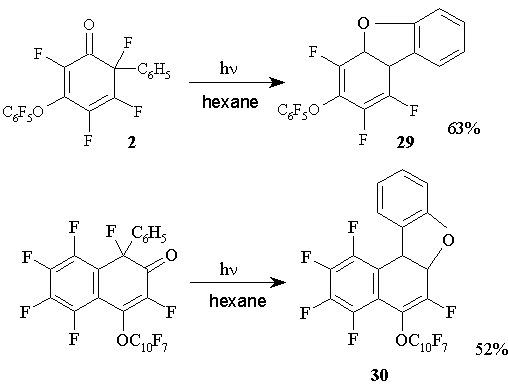
Figure 16
The main product of the photolysis of 2-tolyl-1-oxoheptafluoro-1,2-dihydronaphthalene 31 in chloroform solution is fluorinated derivative of benzonaphtofuran 32.В Moreover, the product of fluorine atom migration that is 2-tolyl-1-oxoheptafluoro-1,4 -dihydronaphthalene 33 is also produced though in small quantity. Besides, for the studied polyfluorocyclohexadienes this is the only reaction where the formation of cyclopropane derivative 6a-tolilheptafluoro-1,1a-dihydro-cyclopropane [a] indenone (34) is observed, typical however, for their hydrocarbon analogues. (Figure 17)
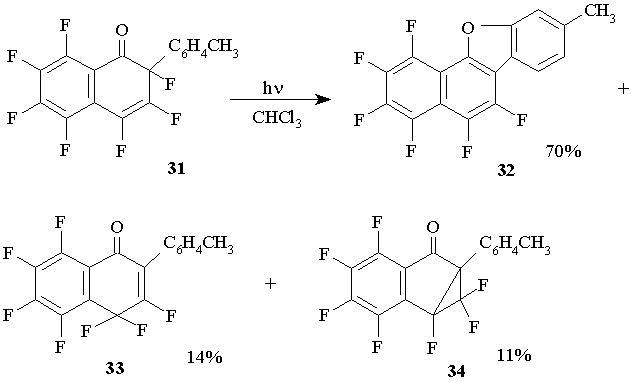
Figure 17
Cycloaddition reactions of polyfluorinated cyclohexadienones
1,3-cycloaddition of diazomethane and its derivatives
Alkyl-substituted 2,4-cyclohexadienoneВ is known to react with diazomethane and alkyl-or phenyldiazomethanes only by their carbon-carbon double bonds, thus forming polycyclic pyrazoles and cyclopropanes [37-40].
The main products of the reaction between cyclohexadienone 14 and phenyldiazomethane in acetonitrile (phenyldiazomethane solution is added to the solution of dienone 14) are isomeric 3-chloro-1,3,4,5,6-penta-fluoro-7-phenylbicyclo-[4.1.0] hepta-4 -en-2-ones (35a and 35b, 62:38), formed with the participation of their double bonds only. In addition, isomeric 6-chloro-3a,4,5,6,7a-penta-fluoro-3,3’-diphenyl-3a,6,7,7a-tetrahydro-spiro[3H-indazol-7,2’-oxiranes] (36a and 36b, 67:33) were obtained with very low yield (5%)
In contrast, 6-chloro-2 ,3,4,5,6-pentafluoro-2 ,4-cyclohexadienone 14 reacts with diazomethane in ether at 0В°C, both by its carbonyl group and by its double bond resulting in a mixture of two isomers (58:42) 37a, b with the yield 52% [41]. (Figure 18)
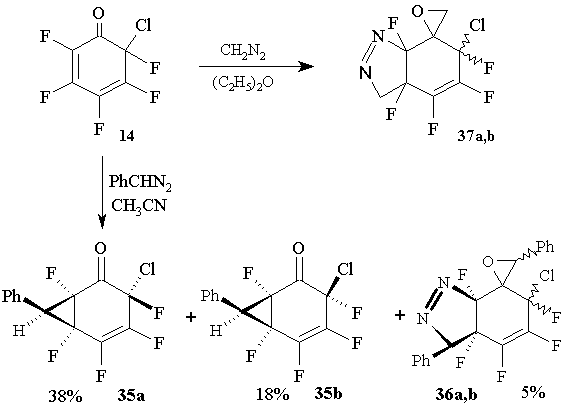
Figure 18
Bicyclotetrahydroindazols of type 36 or 37 were not detected in those reactions. (Figure 19) The reactions of 6-chloro-3-(pentafluorophenoxy)-2,4,5,6-tetrafluoro-2,4-cyclohexadienone 38 or cyclohexadienone 2 with phenyldiazomethane in acetonitrile also result in cyclopropane derivatives 39a and 39b (67:33), or 40a and 40b (83:17), respectively, and in pentafluorobenzylphenyl ether 41 (10%). At the same time (Z) - and (E)-stilbenes 42 are produced in significant amounts being the products of competitive reactions. Bicyclotetrahydroindazols of type 36 or 37 were not found among the reaction products (Figure 19).
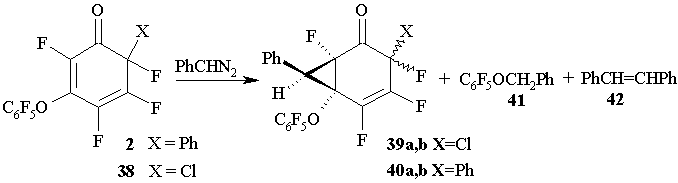
Figure 19
For the formation of cyclopropane derivatives in the reactions of polyfluorinated cyclo-hexadienones 2, 14 and 38 phenyldiazomethane the following mechanism was suggested (Figure 20)
Zwitter-ion 44, formed at the first step from dienones 2, 14 or 38, via a transition state similar to 43, is stabilized either by nitrogen elimination followed by cyclisation, resulting in the formation of cyclopropane derivatives 35a, b, 39a, b and 40a, b or by the elimination of pentafluorophenol, that interacts with phenyldiazomethane resulting in the formation of pentafluorophenylbenzyl ether 41. The probability of the latter reaction was verified in an independent experiment. In addition, it is known that pentafluorophenoxyl group at position 3 of polyfluorinated cyclohexadienone is easily substituted by various nucleophiles [24].

Figure 20
X-ray structure analysis of bicycloheptenons 35a and 35b show that the cycloaddition of cyclohexadienone 14 or phenyldiazomethane occurs with high selectivity: both isomers 35a and 35b have endo-configurations, which is consistent with the above proposed scheme.
It is interesting [42] that the tetrafluoro-n-benzoquinone reacts with diazomethane only by its carbonyl group resulting in tetrafluoro-2 ,5-cyclohexadiene-1-one-4-spiro-2 '-oxirane 45, along with a small amount of pentacyclic compound 46 that is a product of the reaction with the participation of both double bonds and carbonyl groups of tetrafluoro-n-benzoquinone. (Figure 21)
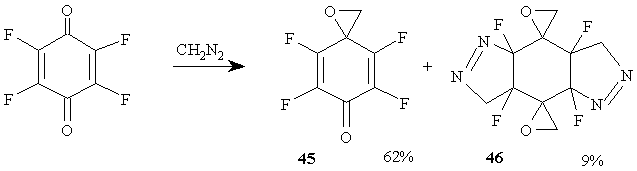
Figure 21
 Polyfluorinated 2,5-cyclohexadienones 8, 47-49, being rather similar to tetrafluoro-n-benzoquinone react with diazomethane by its carbonyl group resulting in high yield (70 – 98%) of a mixture containing two diastereomeric polyfluorinated 1-oxaspiro[2.5]octa-4,7-dienones 50a-52a and 50b-52b, and 53 as well  [43] (Figure 22).

Figure 22
The product of diazomethane addition by the double bond of polyfluorinated 2,5-cyclohexadienone 54 is formed in trace quantity only.
Therefore, polyfluorinated 2,4-cyclohexadienones unlike non-fluorinated cyclohexadienones react with diazomethane both by their double bonds and by carbonyl groups resulting in the formation of isomeric fluorinated tetrahydro-spiro[indazol-oxiranes] that differ in the steric position of halogens in their CFCl-groups. PolyfluorinatedВ 2,4-cyclohexadienones react with phenyldiazomethane mostly by their fluorinated double bonds resulting, with high stereoselectivity, in isomeric fluorinated phenylbicyclo[4.1.0]heptenones in endo-configuration. В While so doing polyfluorinated 2,5-cyclohexadienones react with diazomethane by its В carbonyl group mostly.
For polyfluorinated 2,5-cyclohexadienones the replacement of carbonyl groups for reactive oxirane cycles expands considerably their synthetic potentialities. The addition of alkyl- or arylisocyanates resulting in the formation of 2-oxazolidinones [44, 45] is an interesting and well-known reaction of oxiranes.
Polyfluorinated spiro-oxiranes 50,51 react with aryl- or alkyl- isocyanates in the presence of lithium or sodium salts resulting in the formation of fluorinated dihydro-1,3-benzoxazol-2(3Рќ)-ones 55 [43] (Figure 23).
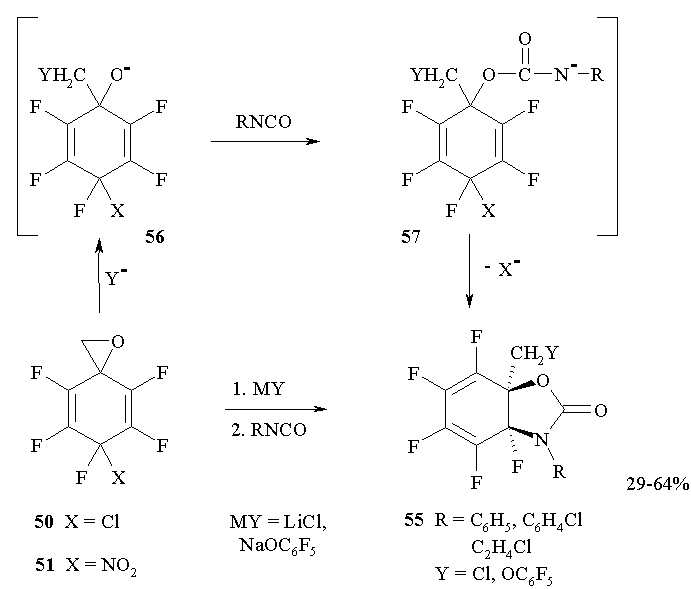
Figure 23
It should be marked that spirooxirane 50 reacts in the presence of catalytic amounts of LiCl, while in the reactions of oxirane 51 lithium or sodium salts perform as reagents and consumed in equimolar quantities. This is in conformity with the assumption about primary cleavage of CH2-O bond under the action of a nucleophile (ClВЇ or ВЇOC6F5), resulting in the formation of alcoholate 56. Addition of anion 56 to isocyanate results in ambident ion 57, that undergoes cycling by РЎ=РЎ bond exclusively followed by the elimination of РҐВЇ from geminal CFX nod and the formation of benzoxazolones 55.
В Some interesting results followed from the efforts to open oxirane cycles in substances 51, 52 under the action of Lewis acids [46, 47]. It is well-known that the interaction between oxiranes and aromatic substances in the presence of Lewis acids results in electrophilic oxyalkylation [48, 49]. We found that the reactions of polyfluorinated spirooxiranes 51,52 with В benzene, toluene, mesitulene, or anisole in the presence of AlCl3 and О±-picoline resulted in the formation of fluorinated biaryls. At the same time oxirane 51 loosing nitrogroup performs as a pentafluorophenylating agent, producing biphenyls 58a-58d. At the same time, oxirane 52, with OC6F5 group in its geminal nod reserves it untouched in the reaction products, therefore resulting in biaryls 59a,b. In fact, in the reactions of electrophilic substitution in aromatic nucleus oxiranes 51, 52 perform as "synthetic equivalent" of polyfluorinated aryl cations. (Figure 24)

Figure 24
В One may assume that complex "A" formed by oxirane (51 or 52) with AlCl3 at the first step is opened with the formation of arenonium ion "B". The latter interacts with aromatic substrate probably via transient zwitter-ion "C" resulting finally in the formation of polyfluorinated biaryls 58a-d, 59a,b, though the possibility of direct interaction between complex "A" and aromatic substrate is not ruled out..
Reactions [4+2] of cycloaddition with acetylene derivatives
Polyfluorinated 2,4-cyclohexadienones easily enter into a reaction of [4+2] cycloaddition with dienophiles such as acetylene derivatives containing aryl- , alkyl-, or hydroxyalkyl groups, resulting in high yield (75-96%) of bicyclo[2,2,2]octadienes 60 [19,50-52]. Those reactions proceed under soft conditions on boiling in benzene, toluene, or carbon tetrachloride. The cycloaddition occurs with high regio- and stereo- specificity resulting in the formation of isomers 60 only, where R1=H for the reactions with non-symmetric acetylenes, the fact being in conformity with the distribution of electronic density within the diene fragment of original 2,4-cyclohexadienone and acetylene (Figure 25)

Figure 25
The results of x-ray structural analysis confirm [52] high stereoselectivity of Diels-Alder reaction of polyfluorinated cyclohexadienones with acetylenes. The configuration of sp3- Sp3-hybridized carbon atom with a fluorine atom and substituent X does not depend on the nature of the said substituent: the fluorine atom is always oriented towards the bond formed due to the dienophile addition. The computation by the molecular mechanical method (MM) [52] of two epimeric adducts with different positions of fluorine and chlorine atoms at sp3-hybridized carbon atom demonstrated that high stereoselectivity of the reaction is due to the thermodynamic stability of thus formed adducts, but is likely a result of less hindered attack by the dienophile on the cyclohexadienone from fluorine side as to compare with those from other geminal substituents, i.e. В chloro-, phenyl-, or pentafluorophenoxyl groups [52].
An interesting result was obtained for the cycloaddition of 3-azidoРѕ-tetrafluoro-6-chloro-2,4-cyclohexadiene-1-on 61. The main product of this dienone heating with phenylacetylene in CCl4 at 70В°C was 4-oxo-2-phenyl-3,5,6,7-tetrafluoro-5-chloro-bicyclo[4.1.0]hepto-2-en-7- carbonitrile 62[19]. Cyclopropane derivative 62 is a product of cycloadduct 63 conversion, its formation being confirmed by 19F NMR spectra during the reaction conducted at room temperature. (Figure 26)

Figure 26
Phosphaalkynes are hetero-analogues of acetylene that also enter the reaction [4+2] of cycloaddition with polyfluorinated cyclohexadienones [53]. Earlier the reactions [4+2] of phosphaalkyne cycloaddition to cyclic 1,3-dienes were applied in the synthesis of cyclic phosphaalkenes, particularly, of phosphoaromatic substances [54, 55]. Usually, the researchers failed to isolate the intermediate bicyclic adducts in those reactions [56].
Heating cyclohexadienone 2 with bis(isopropyl)aminophosphaethine in РЎH2Cl2 solution from -196 В°C to room temperature we succeeded to produce with quantitative yield only one isomeric bicyclic adduct 64 [53]. (Figure 27)

Figure 27
Both chemical composition and structure of adduct 64 were determined by elemental analysis and spectral studies (MS, NMR 1H, 19F, 31P, 13C).
It is interesting that unlike the above reaction the interaction of cyclohexadienone 2 with В tert-butylphosphaethine results in the formation of a tree-component mixture of substances 65a,b and 66 at ratio 90:4:6. The results of 31P NMR spectroscopy allow for concluding that in the product blend of this reaction regioisomer 65 is present in the form of two different stereomers, while regioisomer 66 is present in a single form only. The rate of reaction with tert-butylphosphaethine is markedly less, because the conversion of cyclohexadienone 2 is completed only after two days of heating the reagent mixture at 60 В°C. (Figure 27)
Bicyclic adducts 64, 65a,b and 66 are stable in solid state or in usual organic solvents, and with surplus phosphaalkyne they do not enter Diels–Alder homo-reaction so typical for 2-phosphabicyclo [2.2.2] octa-2, 5-diene [57]. However, in chloroform solution substance 65a undergoes intramolecular [2+2] cycloaddition initiated probably by diffused daylight. Adduct 65а after aging for a week at room temperature is 60% isomerised to substance 67. (Figure 28)

Figure 28
В В The tetracyclic structure of substance 67 was assumed reasoning from the analysis of NMR spectroscopy data. The observed considerable shift towards strong field of signal 31P (Δδp 304.1 m.d.) caused by valent isomerisation of substance 65a (δp = 217.7 m.d.) to 67 (δ p = -86.4 m.d.), is in conformity with the shift value Δδp 445 m.d., observed during the photochemical transformation of 2-dewar-phosphine to corresponding derivative of phosphoprismane [58].
Polyfluorinated cyclohexadienones enter the reactions of [4+2] cycloaddition with such dienophile as dehydrobenzene [59]. The former was produced in situ from Рѕ-aminobenzoic acid and isoamylnitrite [60-62]. (Figure 29)
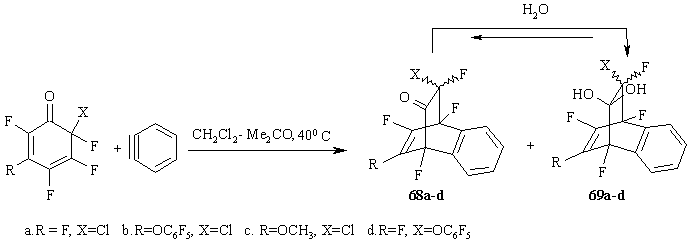
Figure 29
The low-yield (26-37%) reaction products are a mixture of two substances 68a-d and В 69a-d. From the analysis of their NMR19F and NMR 13C spectra one may conclude that substances 69a-d are in fact hydrated cycloadducts 68a-d, formed probably with the participation of water available in the solvent.
Reactions of [4+2] cycloaddition to alkenes
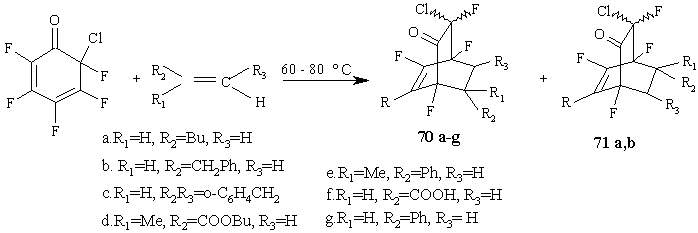
The reactions of polyfluorinated cyclohexadienones with alkenes [63] result in the high-yield (84-97%) formation of bicyclic adducts 70a-f. With most of alkenes those reactions proceed with high regioselectivity, like those with acetylenes [52], except the reactions with 1-hexene or allylbenzene that result in a equal mixture of two isomeric cycloadducts 70a,b and 71a,b, probably in consequence of slight polarization of the double bond in those alkenes. (Figure 30)
В Figure 30
The reactions between cyclohexadienone 14 and a-fluorostyrene 72a or a-fluoro-4-chlorostyrene (72b) in boiling benzene result in the products of [4+2] cycloaddition that is an equal mixture of isomer: endo-73a,b and exo-73a,b [64]. The reaction between cyclohexadienone 2 and a-fluorostyrene 72a proceeds in similar manner though under more drastic conditions (toluene, 110°С), resulting in a couple of isomeric bicyclic adducts (endo-74 and exo-74) in nearly equal proportion.В (Figure 31)
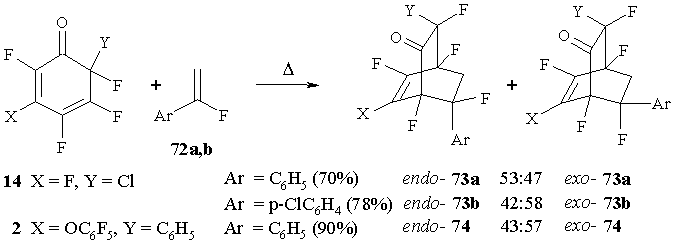
Figure 31
High stereoselectivity is observed in the reactions between cyclohexadienones 2 or В 14 and В a-fluorostyrenes 72a,b, as well as in those with acetylenes: fluorine of CFX group in bicyclo[2.2.2]octenones 73 and 74 is oriented towards the new bond, formed due to the addition of a-fluorostyrene 72a (X-ray analysis of endo-73a, endo- and exo-74) [64].
The attempts to conduct the reaction of 2,4,5,6-tetrafluoro-3-metoxy-6-phenyl-2,4-cyclohexadienone 7 with В О±-fluorostyrene 72a failed. Instead of bicyclic adducts the reaction resulted only in a product of 1,3-shift of fluorine: 2,4,4,5-tetrafluoro-3-metoxy-6-phenyl-2,5-cyclohexa-dienone 75. (Figure 32)
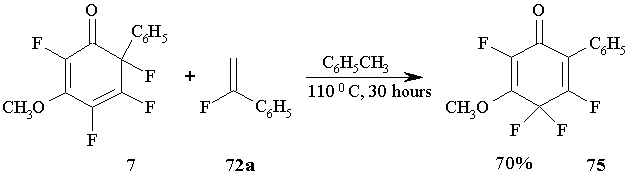
Figure 32
It is well-known that ОІ-fluorostyrene in the reaction with 1,3-diphenylisobenzofuran perform as a dienophile less active than О±-fluorostyrene[65]. The reactions of trans-ОІ-fluorostyrenes 76a-d with polyfluorinated 2,4-cyclohexadienones proceed slower than those with О±-fluorostyrenes. However, boiling of cyclohexadienone 14 with trans-ОІ-fluorostyrenes 76a-d in toluene result in the formation, with yield 75-79%, of two bicyclicВ adducts, those being endo-trans-77 and exo-trans-77 isomers, with predominance of endo-trans-isomers in all cases. (Figure 33)
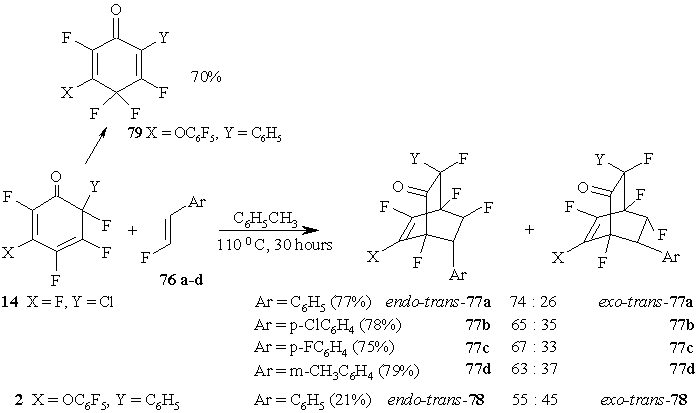
Figure 33
Cyclohexadienone 2 demonstrates low reactivity in the interaction with trans-ОІ-fluorostyrene 76a and it isomerizes predominantly into 2,4,4,5-tetrafluoro-3-(pentafluorophenoxy)-6-phenyl-2,5-cyclohexadienone 79; the products of cycloaddition (endo-trans-78 and exo-trans-78) were produced with small yields. (Figure 33)
Under similar conditions the isomerisation of cyclohexadienone 7 results in one product only 75, as if in the reaction with О±-fluorostyrene. (Figure 32)
Quantum-mechanical calculations [64] using РM3 method [66] are in conformity with the results on the interaction of 2,4-cyclohexadienones with α- or β-fluorostyrenes. The comparison between HOMO and LUMO energies, and between orbital coefficients of cyclohexadienone - styrene pairs that participate in the process, demonstrates that their interaction is an agreed process with reverse electronic request. Thus, for cyclohexadienone 14 - styrene 72a pair the interaction HOMOstyrene - LUMOdienone (О”=6.94 Ev) is more profitable as to compare with HOMOdienone - LUMOstyrene (О”=10.33 Ev). In addition, the orbital coefficients demonstrate very effective overlapping. For reaction between substance 14 and 76a the observed picture is similar: HOMOstyrene - LUMOdienone (О”=6.76 Ev) and HOMOdienone - LUMOstyrene (О”=10.33 Ev). However, in this case the orbital overlapping is less efficient. For the reactions of substance В 2 with В 72a and 76a the differences between HOMOstyrene and LUMOdienone energies are larger (7.30 Ev and 7.12 Ev, correspondingly) and, more than that, in the reactions of substance 2 with 72a the involved orbitals overlap less, and even more less in the reactions of substance 2 with 76a. As a consequence, in this case under thermal conditions partial isomerisation of substance В 2 to 79 is a more probable by-process (see Figure 33).
At last, in the reactions of methoxy-substituted cyclohexadienone 4 with 72a or 76a the difference in the energy of participating orbitals reaches its maximum (7.60 Ev or 7.42 Ev, correspondingly) and cycloaddition at those conditions is not observed, but only the isomerisation of 4 to 75 takes place (see Figure 32).
All attempts to conduct Diels-Alder reaction of polyfluorinated 2,4-cyclohexadienones with the participation of a-fluoro-a,b-unsaturated ketones, e.g., 4-fluoroocto-4-en-3-on, were resultless. At the same time, cyclohexadienone 14 easily enters the reaction of diene synthesis with such dienophile as naphthoquinone, resulting in high yield of stable adduct 80 [67]. (Figure 34)
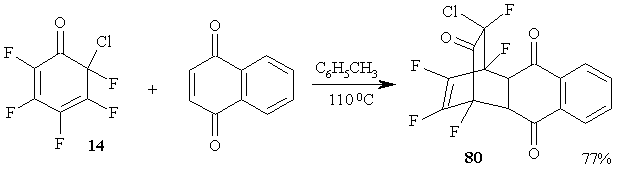
Figure 34
Conversion of fluorinated bicyclo[2.2.2]octadienones
Photochemical reactions
The photolysis of fluorinated bicyclo[2.2.2]octadienones 60, 68a with CFCl group in the position adjacent to carbonyl group at room temperature in chloroform solution results in the formation of, mainly, tetrafluoroaromatic substances 81, probably, due to the elimination of fluorochloroketene (FClC=C=O) [68]. The observed conversion of bicyclo[2.2.2]octadienones is 100-75%, and the yield of fluoroaromatic substances vary between 50 and 86%, as recalculated to non-reacted bicyclo[2.2.2]octadienone. (Figure 35)

Figure 35
Selective reductive dehalogenation of halogen atoms at sp3-hybridized carbon atom
The reaction of bicyclic adducts of 8-chloro-1,2,3,4,8-pentafluoro-bicyclo[2.2.2]octa-2,5-dienones 60 with Zn in acetic acid allows step-by-step replacement of chlorines for hydrogens, and further for fluorines at sp3-hybridized carbon atom [69]. The structure of new bicyclic adducts 82a-d and 83a-d was ascribed to them reasoning from the spectral and analytical data. The fluorines in other positions stay untouched. (Figure 36)
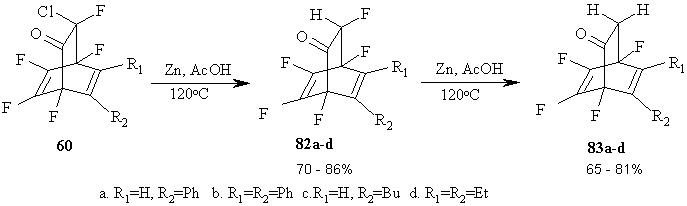
Figure 36
It should be marked that though the replacement of chlorine for hydrogen in a-position to carbonyl group, particularly in cyclic ketones, is well-known [70], but no published data were found about similar replacement of fluorine atoms [71].
Hydrolysis of bicyclic adducts in the presence of various alcohols and amines
One interesting property of bicyclic adducts produced by the reactions of polyfluorinated cyclohexadienones with acetylene derivatives is their easy splitting under the action of nucleophilic reagents with the formation of fluorine-containing derivatives of arylacetic acids 80-83. The peculiarity of those reactions is the retention in those aromatic substances of bridge bond structural elements with CFCl and carbonyl group [51]. Bicyclic adducts resulting from the replacement of geminal chlorines or fluorines for hydrogens 82,83 [69] are easily aromatised with high yield (85-95%) of corresponding fluorinated arylacetic acids 88,89. (Figure 37)
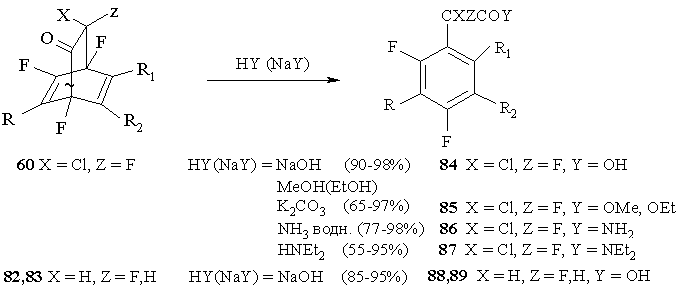
Figure 37
The treatment of cyclic adducts 68a-d produced from polyfluorinated 2,4-cyclohexadienones and dehydrobenzene, with a base or ammonia in water dioxane solution results in the formation of fluorine-containing naphthylacetic acids 90a-d or their amides 91a-d [59] with high yields. It should be marked that in those cases the aromatization occurs with conserving of the structural elements of bridges containing carbonyl groups. (Figure 38)
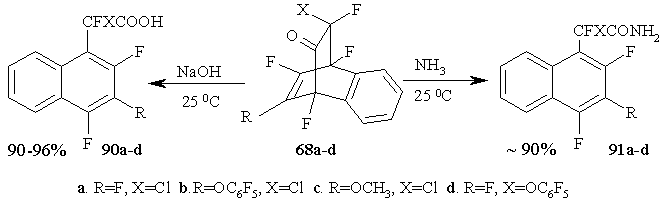
Figure 38
Unlike those reactions some fluorinated or non-fluorinated analogues of the said adducts are aromatised only under the action of strong bases followed by the elimination of the bridge [72] involving a carbonyl group, or they are unclosed with the formation of non-aromatic cyclic В substances [73], or do not react with water base [72].
The mechanism of Diels-Alder aromatisation of the polyfluorinated adducts involves evidently the split of C(4)-C(7) bond (path Рђ) under the action of a nucleophile and the formation of a stable anionic Пѓ-complex 92, that undergoes aromatisation eliminating fluoride-ion from the geminal nod of the cycle. (Figure 39)
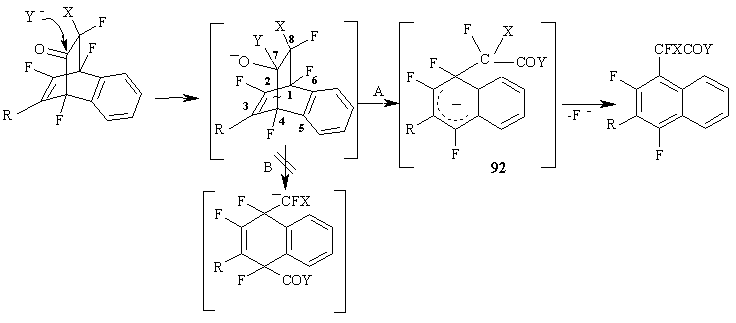
Figure 39
One may see that the alkaline cleavage of bicyclic adducts of polyfluorinated cyclohexadienones produced by Diels-Alder reaction is a convenient method for the synthesis of В fluorinated derivatives of arylacetic acids. With variable substituents in acetylene, in geminal and 3 positions of polyfluorinated 2,4-cyclohexadienone, using different alcohols and amines at the stage of adduct aromatization one may produce various derivatives of arylacetic acids, containing functional groups along with fluorine atoms. It is difficult to produce this type of promising biologically active substances by another method.
Cycloadducts formed in the reactions between polyfluorinated cyclohexadienones and alkenes 70 easily undergo alkaline cleavage in water dioxane solution resulting in fluorine –containing cyclohexenecarbonic acids 93 with rather high yields (65-95%) [63]. The configuration of those substances was confirmed by x-ray structure analysis of two samples of cyclohexenecarbonic acid. (Figure 40)
The most interesting aspect of the reaction is that under the action of hydroxide anion on the adducts of the reactions between polyfluorinated cyclohexadienones and alkenesВ it is bond C(7)-C(8) (path Р’) undergoes cleavage, but not bond C(4)-C(7) (path Рђ), as it has place for bicyclic adducts, formed through the interaction of polyfluorinated cyclohexadienones with acetylenes or dehydrobenzene (see Figure 39). When so doing, the intermediate anion 94 with negatively charged carbon atom linked with fluorine and chlorine is stabilized due to the addition of proton and formation of cyclohexenecarbonic acid [63].

Figure 40
Cycloadduct 80 produced through the interaction of polyfluorinated cyclohexadienone В 14 with 1,4-naphtoquinone under the action of sodium hydroxide at room temperature undergoes the cleavage of C-C bond in its O=C-CFCl fragment and simultaneous elimination of В hydrogen fluoride resulting in the formation of fluorinated anthraquinonecarbonic acid 95. This new method is a convenient way to functional derivatives of fluorinated anthraquinones [67]. (Figure 41)

Figure 41
Conclusion
The synthetic conversions here reviewed are based on the use of polyfluorinated cyclohexadienones for synthons and allow the production by the methods of photolysis, nucleophilic reactions, cycloaddition and simple conversion of those reactions products a wide range of fluorinated substances: derivatives of polyphenyl ethers, biphenyls, arylacetic acids, derivatives of naphthalene or anthraquinone, containing carboxyl groups, cyclohexenecarbonic acids, fluorinated heterocyclic substances, cyclopropane derivatives and bicyclic phosphorus-containing substances. The further transformation and modification of those reactions products make it possible to manufacture various structured organofluoric substances earlier inaccessible.
The new approach is particularly important as a method for the synthesis of fluorinated derivatives of aryl- or diarylacetic acids, because the representatives of this substance category provide the basis for many drugs (propanidid, diclofenac, spazmolitin, pafentsil, aprofen). It is well-known that the presence of fluorine atoms in pharmaceutical preparations enforces their activity, improves their metabolic stability, prolongs their active life time, and frequently decreases their toxicity [74-76].
The approach in what polyfluorinated cyclohexadienones may be used for highly reactive synthons, opens promising perspectives for the synthesis of earlier inaccessible physiologically active fluorinated arylacetic acids that contain functional groups. Those synthetic processes are realizable due to the possibility of introduction of the required functional groups at any step of the process of the acid synthesis: nucleophilic substitution of fluorine atom t position 3 of В polyfluorinated cyclohexadienones; using of various substituted acetylenes in the reactions of cycloaddition; selective replacement of halogens for hydrogen atoms at sp3-hybridized carbon atom in bicyclic adducts; hydrolysis of bicyclic adducts with various alcohols or amines resulting in the formation of derivatives of arylacetic acids.
References
2. Kobrina L.S.// Izv. AN. Ser. khim. 2002. s. 1629-1649
3. Ahmetova N.E., Shtark A.A., Shteingarts V.D.// Zhurn. org. khimii. 1973. T. 9. s 1218- 1227
4. Kobrina L.S., Kovtonyuk V.N., Yakobson G.G.// Zhurn. org. khimii. 1977. T. 13. s. 1447- 1452
5. Kovtonyuk V.N., Kobrina L.S., Yakobson G.G.// Izv. SO AN SSSR, Ser. khim. nauk. 1984. vyp. 2. № 5. s. 119-123
6. Kovtonyuk V.N., Kobrina L.S., Yakobson G.G.// J. Fluor.Chem. 1981. V. 18. P. 587-600
7. Kovtonyuk V.N., Kobrina L.S., Yakobson G.G.// J. Fluor.Chem. 1985. V. 28. P. 89-98
8. Kobrina L.S., Popkova N.V., Yakobson G.G.// Izv. SO AN SSSR, Ser. khim. nauk. 1976. vyp. 3. № 7. s. 125-128
9. Kobrina L.S., Popkova N.V., Yakobson G.G.// Izv. SO AN SSSR, Ser. khim. nauk. 1976. vyp. 5. № 12. s. 140-147
10. Popkova N.V., Kobrina L.S., Yakobson G.G. //Izv. SO AN SSSR, Ser. khim. nauk. 1978. vyp. 5. № 12. s. 110-116
11. Kovtonyuk V.N., Kobrina L.S.// J. Fluor. Chem. 1993. V. 63. P. 243-251
12. Miller B.// J. Org. Chem. 1963. V. 28 P. 345-348
13. Pat. 3525756 US // Chem. Abstrs. 1970. V. 73. P. 98477
14. Sheppard W., Sharts C.// Organic Fluorine Chemistry. New-York : W. A. Benjamin, Inc. 1969
15. Somekawa K., Matsuo T., Kumamoto S.// Bull. Chem. Soc. Jpn. 1969. V. 42. P. 3499-3504
16. Ahmetova N.E., Kostina N.G., Mamatyuk V.I., Shtark A.A. Shteingarts V.D.// Izv. SO AN SSSR, Ser. khim. nauk. 1973. vyp. 6. № 14.s. 86-100
17. Ahmetova N.E., Kostina N.G., Shteingarts V.D. //Zhurn. org. khimii. 1979. T. 15. s. 2137-2147
18. Kovtonyuk V.N., Kobrina L.S., Yakobson G.G.// Zhurn. org. kimii. 1988. T. 24. s. 1952- 1959
19. Kovtonyuk V.N., Kobrina L.S.// Zhurn. org. khimii. 2002. T. 38. s. 196-201
20. Ahmetova N.E., Shteingarts V.D.// Zhurn. org. khimii. 1977. T. 13. s. 1277-1285
21. Kovtonyuk V.N., Kobrina L.S.// Zhurn. org. khimii. 1991. T. 27. s. 2289-2297
22. Kovtonyuk V.N., Kobrina L.S.// ZHurn. org. himii. 1999. T. 35. S. 82-86
23. Kovtonyuk V.N., Kobrina L.S., Yakobson G.G.// Zhurn. org. khimii. 1979. T. 15. s. 1447- 1453 2
24. Grobe J., Le Van D., Wiese H., Krebs B., Läge M., Kobrina L.S.// Z. Naturforschung B. 1995. V. 50. P. 691-694
25. Waring A.J.// Advances in Alicyclic Chemistry. (Eds. Hart H. and Karabatsos G.J.). New York: Acad.Press. 1966. V. 1. 129 P.
26. Taylor E.C., Jagdmann G.E., McKillop A.// J. Org. Chem. 1978. V. 43. P. 4385-4387
27. Auwers K., Ziegler K.// Liebigs Ann. Chem. 1921. V. 425. P. 280-294
28. Bamberger E., Reber E.// Ber. Deutsch. Chem. Ges. 1907. V. 40. P. 2258-2274
29. Hecker E.// Chem. Ber. 1959. V. 92. P. 3198-3208
30. Kovtonyuk V.N., Kobrina L.S.// Izv. AN. Ser. khim. 1996. s. 1778-1781
31. Gozzi F., Siegel J.S.// Pure Appl. Chem. 1995. V. 67. P. 683-689
32. Perst H., Dimroth K.// Tetrahedron. 1968. V. 24. P. 5385-5393
33. Greenland H., Pinhey J.T., Sternhell S.// J. Chem. Soc., Perkin Trans. 1. 1986. P. 1789-1795
34. Greenland H., Kozyrod R.P., J.T.Pinhey J.T.// J. Chem. Soc., Perkin Trans. 1. 1986. P. 2011- 2016
35. Carnduff J., Iball J., Leppard D.G., Low J.N.// J. Chem. Soc., Chem. Commun. 1969. P. 1218-1219
36. Kovtonyuk V.N., Kobrina L.S.// J. Fluor. Chem. 1994. V. 66. P. 219-221
37. Wessely F., Schinzel E., Spiteller G., Klezl P.// Monatsh. Chem. 1959. V. 90. P. 96-117
38. Spiteller G., Schmidt G., Budzikiewicz H., Wessely F.// Monatsh. Chem. 1960. V. 91. P. 129-138
39. Wessely F., Budzikiewicz H., Metlesics W.// Monatsh. Chem. 1959. V. 90. P. 121-133
40. Wessely F., Budzikiewicz H., Janda H.// Monatsh. Chem. 1960. V. 91. P. 456-464
41. Kovtonyuk V.N., Kobrina L.S., Gatilov Yu.V., Bagryanskaya I.Yu., Frölich R., Haufe G.// J. Chem. Soc., Perkin Trans. I. 2000. P. 1929-1933
42. Kovtonyuk V.N., Kobrina L.S., Bagryanskaya I.Yu., Gatilov Yu.V.// Zhurn. org. khimii. 1999. T. 35. s. 75-78
43. Kovtonyuk V.N., Kobrina L.S.,О.M.Kataeva, G.Haufe.// Eur. J. Org. Chem. 2005. P. 1178- 1183
44. Dyen M.E., Swern D.// Chem. Rev. 1967. V. 67. P. 197-246
45. Okumoto S., Yamabe S.// J. Comput. Chem. 2001. V. 22. 316-328
46. Kovtonyuk V.N., Kobrina L.S., Petrov D.V. Haufe G.// Tezisy 7-j Vserossijskoj konferencii «Khimiya ftora». Moskva. 2006. R. 148
47. Kovtonyuk V.N., Kobrina L.S., Haufe G.// Izv. AN. Ser. khim. 2008. s. 1654-1656
48. Shorygina N.V.// Zhurn. obshch. khimii.1951. T. 21. s. 1273-1276
49. Hopff H., Koulen K.// Chem. Ber. 1952.V. 85. P. 897-900 3
50. Kobrina L.S., Shteingarts V.D.// Zhurn. org. khimii. 1988. T. 24. s. 1344-1345
51. Kobrina L.S., Bogachev A.A.// J. Fluor. Chem. 1993. V. 62. P. 243-258
52. Bogachev A.A., Bagryanskaya I.Yu., Rybalova T.V., Gatilov Yu.V., Kobrina L.S.// Zhurn. obshch. khimii. 1996. T. 66. s. 1324-1333
53. Grobe J., Le Van D., Broschk B., Kobrina L.S.// Tetrahedron Lett. 1993. V. 34. P. 4619-4622
54. Regitz M. In Multiple Bonds and Low Coordination in Phosphorus Chemistry. (Eds., Regitz M., Scherer O.J.). Stuttgart, New York: G.Thieme Verlag. 1990. P. 58
55. Märkl G., Jin Yu G., Silbereisen E.// Angew. Chem. Int. Ed. Engl. 1982. V. 21. P. 370-371
56. Annen U., Regitz M., Klugeb H.// Chem. Ber. 1990. V. 123. P. 935-937
57. Annen U., Regitz M.// Tetrahedron Lett. 1988. V. 29. P. 1681-1684
58. Blatter K., Rösch W., Vogelbacher U.J., Fink J., Regitz M.// Angew. Chem. Int. Ed. Engl. 1987. V. 26. P. 85-86
59. Bogachev A.A., Kobrina L.S., Shteingarts V.D.// Izv. AN. Ser. khim. 1994. s. 1634-1637
60. Del Mazza D., Reinecke M.C.// J. Org. Chem. 1988. V. 53. P. 5799-5805
61. Friedman L., Logullo F.M.// J. Am. Chem. Soc. 1963. V. 85. P. 1549
62. Gray A.C.G., Hart H.// J. Am. Chem. Soc. 1968.V. 90. P. 2569-2578
63. Bogachev A.A., Manujlov A.V., Kobrina L.S.// Zhurn. org. khimii. 1996. T. 32. s. 1016- 1021
64. Bogachev A.A., Kobrina L.S., Meyer O.G.L., Haufe G.// J. Fluor. Chem. 1998. V. 97. P. 135- 143
65. Ernet T., Haufe G.// Tetrahedron Lett. 1996. V. 37. P. 7251-7252
66. Stewart J.J.P.// MOPAC93. Tokyo: Fujitsu Limited. 1993. 414P
67. Bogachev A.A., Kobrina L.S.// Zhurn. org. khimii. 1997. T. 33. s. 1433-1434
68. Bogachev A.A., Kobrina L.S.// Zhurn. org. khimii. 1997. T. 33. s. 742-744
69. Bogachev A.A., Kobrina L.S.// J. Fluor. Chem. 1998. V. 92. P. 33-39
70. Brady W.T.// Tetrahedron. 1981. V. 37. P. 2949-2966
71. De Kimpe N., Verhe R.// The Chemistry ofa-Haloketones, a-Haloaldehydes and aHaloimines. Chichester: Wiley. 1988. 107P
72. Oku A., Matsui A.// Bull. Chem. Soc. Jpn. 1977.V. 50. P. 3338-3343
73. Mihajlova I.F., Barhash V.A.// Zhurn. org. khimii. 1970. T. 6. s. 2325-2330
74. Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications. (Eds., Filler R., Kobayashi Y., Yagupolskii L.M.). Amsterdam: Elsevier. 1993. 386P
75. Biomedical Frontiers of Fluorine Chemistry. (Eds., Ojima I., McCarthy J.R., Welch J.T.). Washington: ACS Symposium Series 639. 1996. 356P
76. Mashkovskij M.D.// Lekarstvennye sredstva.T 1. Har'kov: Torsing. 1998. s. 543
Recommended for publication by Prof. V.E. Platonov
Fluorine Notes, 2012, 84, 1-2
