Received: июнь 2011
DOI 547.29 + 547.321 + 547.722 + 542.91 + 542.952.1 + 546.16 + 547.151
Fluorine Notes, 2012, 82, 1-2
Превращения некоторых перфторолефинов под действием нуклеофилов как способ утилизации отходов промышленности фторорганических материалов
В.В. Бардин
Учреждение Российской академии наук Новосибирский институт органической химии им. Н.Н.Ворожцова
Сибирского отделения РАН, 630090, Новосибирск, пр. Лаврентьева, 9.
e-mail: bardin@nioch.nsc.ru
Аннотация. Рассмотрены способы превращения побочных продуктов фторорганической промышленности (перфторированные алкены, циклоалкены и азаалкены) в практически полезные промежуточные продукты - полифторированные эфиры, амины и гетероциклические соединения со фторсодержащими заместителями.
Ключевые слова: Перфторалкены. Нуклеофильное замещение. Гетероциклические соединения. Перфторированные алкильные заместители.
Оглавление
1. Введение
2. Реакции нуклеофильного замещения и присоединения
3. Синтез гетероциклических соединений с перфторалкильными заместителями
1. Введение
В промышленном производстве перфторированных полимерных материалов, смазок и технических жидкостей и т.п. получаются жидкие отходы, которые состоят главным образом из низкомолекулярных олигомеров. Эти побочные продукты не имеют самостоятельного применения, их трудно утилизировать обычными способами, но они могут служить сырьём для производства других полезных фторорганических продуктов. Одним из направлений их использования является превращение в частично фторированные соединения взаимодействием с нуклеофилами и последующее исчерпывающее фторирование химическим или электрохимическим способом. Выходы целевых продуктов превышают 70-80%, тогда как выходы при прямом фторировании углеводородного сырья редко бывают выше 10-15% [1]. Другим направлением утилизации является превращение в гетероциклические соединения с перфторированными алкильными группами. Такие продукты могут служить сырьём для дальнейшего фторирования в перфторированные насыщенные гетероциклы с атомами азота и/или кислорода в кольце и для получения препаратов медицинского, ветеринарного и сельскохозяйственного назначения [2-6]. Поскольку проблема имеет промышленную, экологическую и конечно, коммерческую составляющие, опубликовано много научных статей и патентов. В данном обзоре приведены лишь наиболее типичные процессы с использованием простых исходных соединений, что позволяет рассматривать его как введение в эту область. В качестве объектов выбраны димеры перфторпропилена (CF3)2CFCF=CFCF3, (CF3)2C=CFC2F5 , перфторалкоксиалкены C3F7OCF=CF2 и C3F7OCF(CF3)CF2OCF=CF2 (производные димера и тримера окиси перфторпропилена), перфтор-1-этилциклогексен (отход производства хромина) и C4F9N=CFC3F7 (производимый из перфтортрибутиламина).
2. Реакции нуклеофильного замещения и присоединения
Фторангидриды C3F7O[CF(CF3)CF2O]nCF(CF3)COF являются олигомерами окиси перфторпропилена и в небольшом количестве получаются при его полимеризации. Нагревание их раствора в полярном апротонном растворителе (обычно диглим или триглим) с содой дает перфторированные алкилвиниловые эфиры C3F7O[CF(CF3)CF2O]nCF=CF2, которые легко выделяются из реакционной смеси. Перфторалкоксиэтилены C3F7OCF=CF2 (1) и C3F7OCF(CF3)CF2OCF=CF2 (2) реагируют с алифатическими спиртами ROH в среде ТГФ, диоксана, ацетонитрила или со смесью ТГФ-ROH в присутствии основания, образуя полифторированные диалкиловые эфиры. В качестве основания могут выступать KOH или соответствующий алкоголят натрия, но триэтиламин оказался неэффективен.
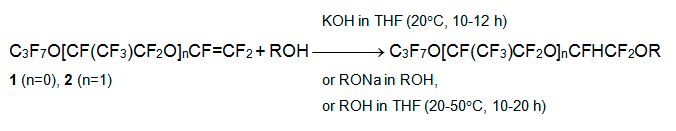
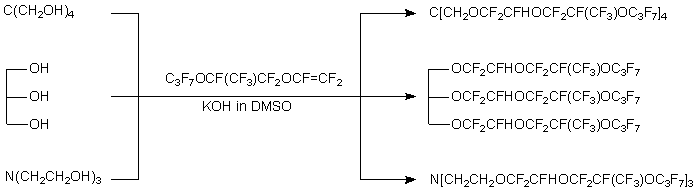
В реакцию вступают алифатические спирты нормального строения (R = CH3, C2H5, C3H7, C4H9), изопропиловый и изобутиловый спирты, метил- и этилцеллозольвы и даже низконуклеофильные фторированные спирты CF3CH2OH, C6F5CH2CH2OH и C6F5OCH2CH2OH [7, 8]. Аналогично присоединяются по С=С связи фенолы 4-R'C6H4OH (R' = CH3OCH2CH2, CH3OC(O)CH2CH2) и C6F5OH. При наличии нескольких гидроксильных групп все они последовательно вовлекаются в реакцию присоединения. Так были получены полифторированные диалкиловые эфиры из многоосновных спиртов (этиленгликоль, триэтиленгликоль, глицерин, пентаэритрит), триэтаноламина и S(CH2CH2OH)2 [7-9].
Для сравнения укажем, что перфторпропилен реагирует со спиртами ROH (R = Me, Et, Pri, CHF2CF2CH2) в присутствии KOH или RONa, давая наряду с полифтордиалкиловыми эфирами немного алкоксипентафторпропилена CF3CF=CFOR [10].
В отличие от О-нуклеофилов, присоединение аминов по С=С связи перфторалкоксиэтиленов протекает без добавки основания. Алкилвиниловые эфиры 1 и 2 реагируют с эквимолярным количеством диалкиламина HNAlk2, пирролидина, пиперидина или морфолина при 25°С, давая (2-полифторалкоксиэтил)амины, которые выделяются перегонкой. Если реакцию проводят в присутствии триэтиламина и конечную реакционную смесь обрабатывают водой, получают диалкиламиды 1-H-перфторалкоксиуксусной кислоты [9, 11].
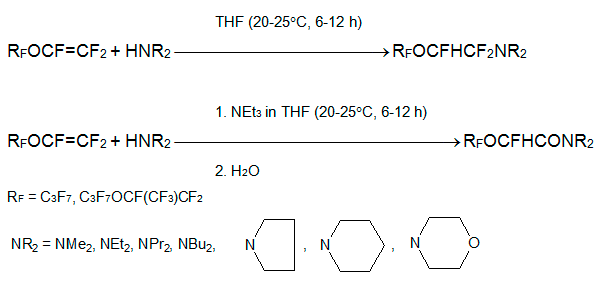
В тех же условиях из эфиров 1, 2 и первичных аминов H2NAlk с хорошими выходами получаются имины RFOCFHCF=NAlk (выделение перегонкой) или алкиламиды RFOCFHCONHAlk (после обработки водой) [9, 11].
В отличие от терминальных перфторалкенов, к которым относятся перфторпропилен и перфторалкоксиэтилены 1 и 2, реакционная способность интернальных перфторалкенов R1CF=CFR2 и R1R2C=CFR3 определяется совокупным электронным и стерическим эффектом атомов фтора при С=С связи и перфторалкильных групп R1, R2 и R3. Это в полной мере относится к реакциям с нуклеофилами. Присоединение нуклеофила к sp2-гибридизованному атому углерода дает карбанион, судьба которого определяется характером заместителей и природой (основностью, полярностью) среды. При этом нуклеофилы могут присоединяться к sp2-гибридизованным атомам углерода и к соседним атомам углерода (перенос реакционного центра). Это порождает разнообразие возможных продуктов, которые к тому же могут претерпевать вторичные превращения. Более подробно реакции интернальных перфторалкенов рассматриваются в обзоре [12], а здесь обобщены результаты нуклеофильного присоединения (замещения) O-, N-, S- и P-нуклеофилов к перфтор-2-метил-2-пентену (3), перфтор-4-метил-2-пентену (4), перфтор-1-этилциклогексену (5) и перфтор-4-аза-5-нонену (6).
Ранее сообщалось, при взаимодействии алкена 3 с метанолом в присутствии триэтиламина происходит замещение атома фтора при кратной связи на метоксильную группу, присоединение метилового спирта по кратной связи (общий выход продуктов 44% в мольном соотношении 1:1) и получается немного изомерных 1,3-диметоксиперфтор-2-метил-1-пентенов [13, 14]. Алкен 4 реагирует только с метилатом натрия, давая изомерные 1,3,4-триметоксиперфтор-2-метил-1-пентены (выход 57%), а образования монометокси продуктов зафиксировано не было [13]. С целью получения сырья для производства перфтор-2-метил-3-метоксипентана газофазным фторированием, было проведено подробное изучение реакций перфторалкенов 3 и 4 c MeOH в разных условиях. Выявлено существенное влияние растворителя (MeOH, DMF, MeCN, с добавками ТГФ или ацетона), соотношение реагентов, порядок смешивания реагентов и природа основания (MeONa, KOH, NEt3). Ниже приведены оптимальные условия и основные продукты реакций [15].
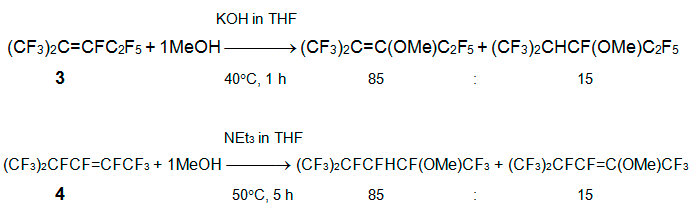
С α,α,ω-тригидроперфторалканолами (спирты-теломеры) перфтор-2-метил-2-пентен реагирует в присутствии триэтиламина или пиридина в MeCN при 70°C, давая только продукты алкоксидефторирования. Более эффективным основанием оказался фторид цезия, который эффективно действует уже при 20°С [16].
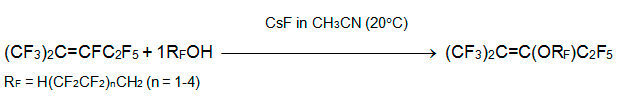
Следует отметить эффективность фторида цезия в реакциях спиртов-теломеров с полифторароматическими соединениями, в которых получаются полифторированные арилалкиловые эфиры [16, 17]. Наряду с алкенилалкиловыми и диалкиловыми эфирами, последние были использованы как сырьё для получения перфтордиалкиловых эфиров путём газофазного фторирования.
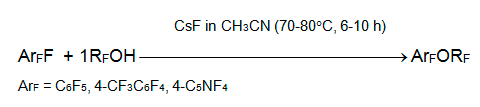
В реакции перфтор-2-метил-2-пентена с моно-, ди- и триэтиленгликолями в соотношении реагентов 3:1 в ацетонитриле (ТГФ, ацетоне, ДМФА) в присутствии триэтиламина (или КОН) алкоксид присоединяется по атому углерода кратной связи с образованием карбаниона, который стабилизируется элиминированием фторид аниона с образованием соответствующих частично фторированных диэфиров. Выходы повышаются при использовании триметилсилиловых эфиров этиленгликолей в безводном MeCN и в присутствии CsF [18].
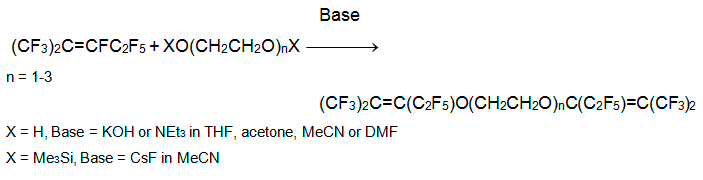
При взаимодействии эквимолярных количеств алкена 3 и этиленгликоля (или в избытке последнего) в присутствии NEt3 получается сложная смесь производных диоксолана и диоксепина.
Фенол и его производные, содержащие электронодонорные заместители, реагируют с перфтор-2-метил-2-пентеном в ТГФ при 40°С, образуя исключительно 2-арилоксипентафтор-2-метил-2-пентены. Аналогично происходит реакция с 2-гидроксибензимидазолом и 2-гидроксибензотиазолом [19].
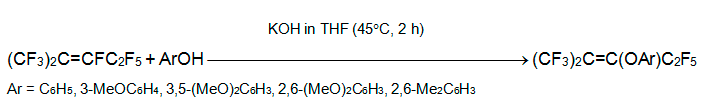
В то же время фенолы с электроотрицательными заместителями в арильном кольце дают смесь 2-арилоксипентафтор-2-метил-2-пентена с 1-арилоксипентафтор-2-метил-2-пентеном. Доля последнего изменяется от нескольких % (Ar = 3-NO2C6H4, 4-NO2C6H4, 3-C5NH4) до 100 (Ar = 2,6-Cl2C6H3, C6F5) [19].
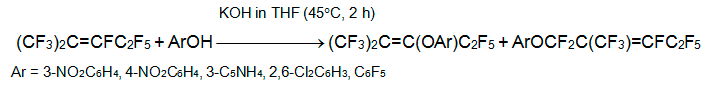
Ранее было показано, что в присутствии триэтиламина алкен 3 образует с оксимом ацетона продукты замещения атома фтора при С=С связи и присоединения к ней [20]. Напротив, оксим ацетофенона реагирует с алкеном 3 в отсутствии основания и дает только продукт присоединения. Дегидрофторирование последнего основанием (триэтиламин) равно и проведение реакции 3 с оксимом ацетофенона в присутствии NEt3 приводят к продукту замещения атома фтора при С=С связи [21].
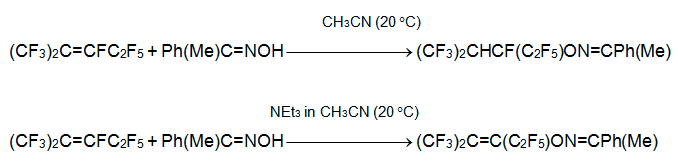
Использование в качестве нуклеофила триметилсилилового эфира оксима ацетофенона приводит сначала к продукту присоединения, который может реагировать с избытком нуклеофила по терминальному атому углерода.
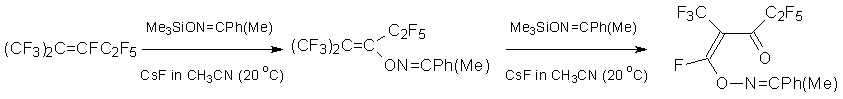
Интересно, что нагревание алкена 3 с гексаметилдисилоксаном и CsF в ДМФА дает эфир енола, который при гидролизе превращается в кетон [22].
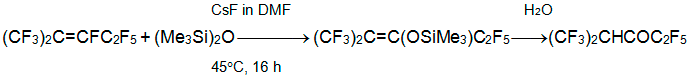
Если О-нуклеофилы дают с алкеном 3 продукты замещения атома фтора при углероде С-3, то реакции N-нуклеофилов характеризуются большим разнообразием продуктов. Так, азид анион вступает в реакцию с алкеном 3 уже при –20°С, образуя термически неустойчивый перфтор-2-метил-3-азидо-2-пентен [23].
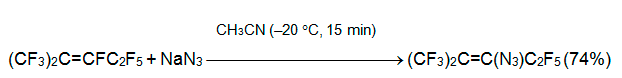
Амбидентный роданид анион менее нуклеофилен, но и в этом случае не требуется добавка основания. Роданид натрия (калия) реагирует с интернальными перфторалкенами 3, 5 и азаалкеном 6 в полярных апротонных растворителях (ацетонитрил, ацетон, ТГФ, ДМФА, сульфолан, ДМЕ) при 40-60°С исключительно с замещением винильного атома фтора [24, 25].
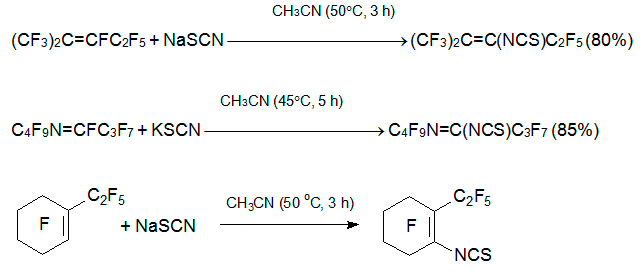
Азолы реагируют с алкеном 3 в присутствии NEt3 подобно O-нуклеофилам, давая гидролитически устойчивые енамины [26]. Качественно аналогичные продукты получаются при кипячении 3 с амидами (пирролидоном, сукцинимидом, фталимидом) в присутствии триэтиламина [27].
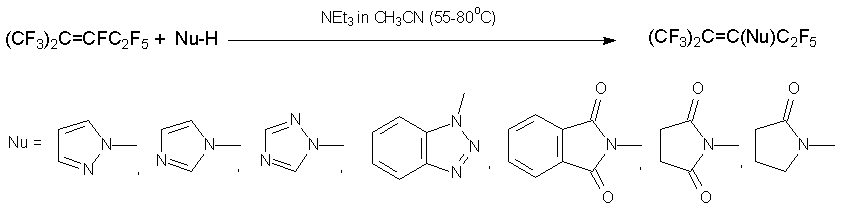
С переходом к более основным вторичным аминам становится существенным другое направление реакции, замещение атома фтора в положении 1. Это обусловлено возрастанием вклада высокоэлектрофильного изомера CF2=C(CF3)CF2C2F5, находящегося в равновесии с изомерами 3 и 4. Так, в реакции с пиперидином, морфолином или бис(4-гептафтортолил)амином соотношение изомерных продуктов A:B составляет 1:3 [27], а в реакции с дипропиламином, дибутиламином и диаллиламином получаются только енамины типа B [28].
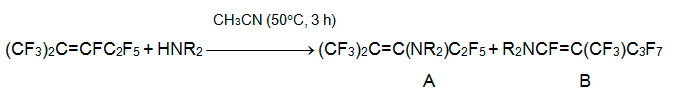
Взаимодействие алкена 3 с гексаметилсилазаном в присутствии оснований начинается атакой N-аниона на атом С-3 и генерированием карбаниона [(CF3)2C-CF(C2F5)N(SiMe3)2]–. В результате последующих фторид-катализируемых реакций получаются 2-Н-перфторизогексан, бис(перфтор-2-метилпент-2-ен-3-ил)амин и 2-Н-3-иминоперфтор-2-метилпентан. Последний является также основным продуктом реакции алкена 3 с безводным NH3 в ТГФ при 0°С, тогда как при комнатной температуре происходят более глубокие процессы [22].
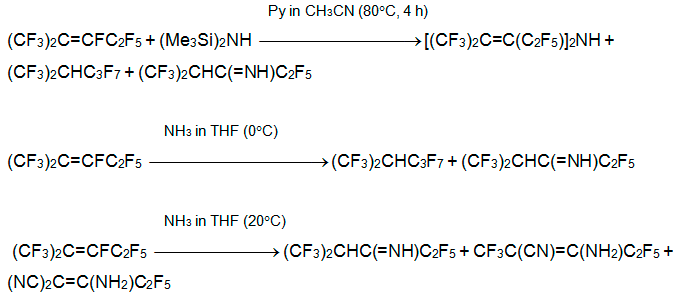
Направление реакции с первичными аминами H2NR существенно зависит от природы R, соотношения алкен:амин и основности среды. Продуктами взаимодействия алкена 3 с первичными алкиламинами, в том числе пространственно затрудненными, являются имины и немного (<10%) производных азетина и азетидина (см. ниже) [29-31].
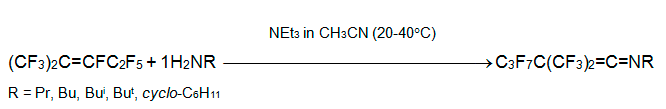
Совершенно другим путем реагирует с интернальными перфторалкенами Р-нуклеофил, триэтилфосфит. При смешивании алкена 3 с 2-мя эквивалентами P(OEt)3 быстро происходит экзотермическая реакция и получается диэтиловый эфир перфтор-2-метилпента-1,3-диен-3-илфосфоновой кислоты. Однако в реакции с перфтор-1-этилциклогексеном родственный эфир фосфоновой кислоты является минорным компонентом, а основным - 1-этилперфтор-2-этилциклогексен. В тех же условиях из перфтор-5-аза-4-нонена и перфтор-1-азациклогексена получаются сложные смеси [32].
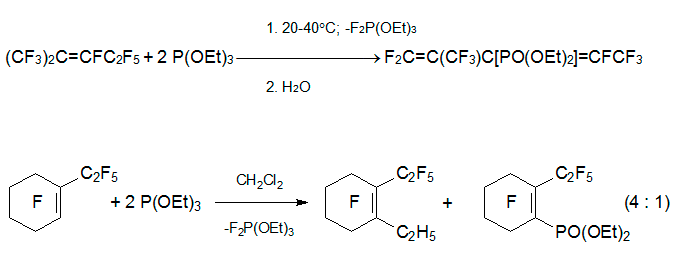
Столь большие отличия в характере продуктов реакции интернальных перфторалкенов с триэтилфосфитом от таковых в реакциях с O- и N-нуклеофилами объясняются сравнительной устойчивостью промежуточных фторфосфоранов и протеканием свойственных им перегруппировок, включающих миграцию атома фтора.
3. Синтез гетероциклических соединений с перфторалкильными заместителями
Наличие нескольких путей стабилизации полифторированного карбаниона, образующегося при присоединения N-нуклеофила к интернальному алкену 3 (CF3)2C=CFC2F5 или азалкену 6 C4F9N=CFC3F7, в ряде случаев приводит к азотсодержащим гетероциклическим соединениям. Так, обработка алкена 3 азидом натрия в смеси MeCN-EtOH при –10°С дает производное 1,2,3-триазолина, а та же реакция в MeCN при 20°С приводит в основном к производному азирина [23].
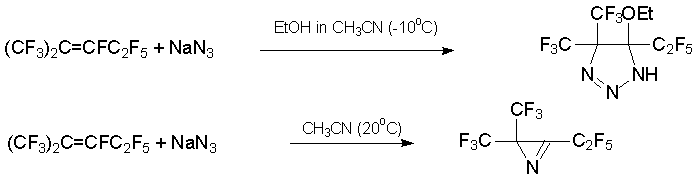
Выше отмечалось, что взаимодействие первичных аминов с алкеном 3 в соотношении 2:1 ведет к иминами. Однако в присутствии триэтиламина основными продуктами становятся производные 4-алкилимино-2-азетина. Вероятно им предшествуют 4,4-дифтор-2-азетины, которые в некоторых случаях удалось выделить из продуктов реакции [29, 30].
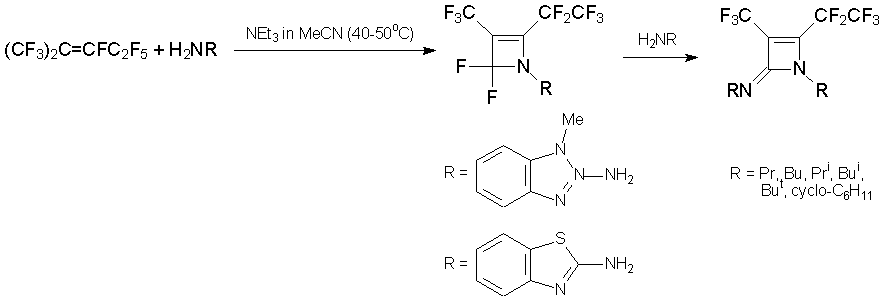
Из азалкена 6 и первичных аминов в близких условиях получаются производные диазетина и перфторбутирамид [31, 33].
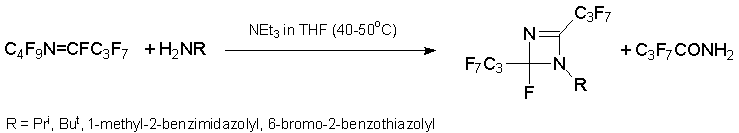
Особо следует отметить реакции с производными анилина. С низконуклеофильными анилинами ArNH2 азаалкен 6 дает производные диазетина (Ar = C6F5, 2-NO2C6H4, 4-NO2C6H4) или азетина (Ar = 2,6-Cl2C6H3), тогда как с высоконуклеофильными анилинами (Ar = C6H5, 4-MeOC6H4, 4-FC6H4, 2,6-(CH3)2C6H3) образуются производные 1,2-дигидрохиназалина. Из алкена 3 в тех же условиях получены производные хинолина [34].
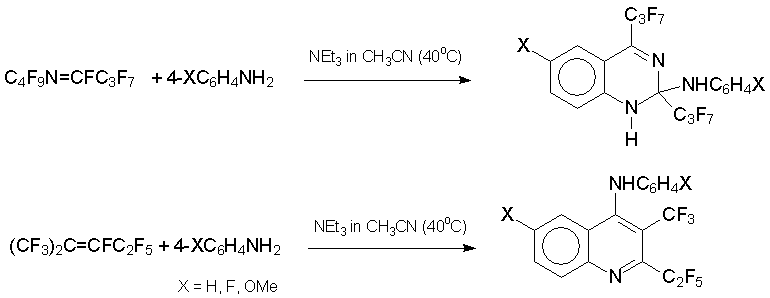
Другой вариант сборки гетероциклов с перфторалкильными группами – реакция интернальных перфторалкенов с бифункциональными нуклеофилами. Так были получены кислород-, азот- и серусодержащие гетероциклы с двумя гетероатомами (из алкена 3), тремя гетероатомами (из азалкена 6) и с разным размером цикла. Эффективность этого метода продемонстрирована синтезом четырёх-, пяти-, шести-, семи- и девятичленных гетероциклических систем. Все реакции протекают в среде MeCN или ТГФ в присутствии триэтиламина при 20-50°С в течение 2-5 часов. Ниже приведены типичные примеры таких синтезов.
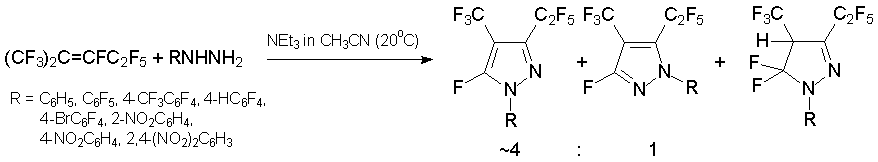
Примером 1N,2N-бинуклеофила являются производные гидразина. Эти соединения реагируют с алкеном 3 образуя (трифторметил)(пентафторэтил)фтор-1-арилпиразолы и немного соответствующего пиразолина [33, 35, 36].
Из азалкена 6 в тех же условиях с выходом 40-70% получаются 3,5-бис(гептафторпропил)-1,2,4-триазолы.
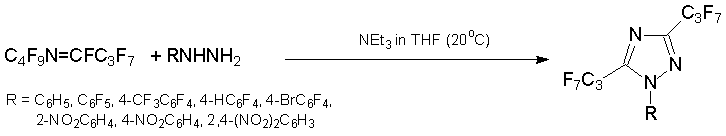
2-Меркаптобензимидазолы [37], 1,2,4-триазол-3-тиол, 1,4,5,6-тетрагидропиримидин-2-тиол [38] относятся к 1N,3S-бинуклеофилам, из которых получаются пятичленные N,S гетероциклы с экзо-тетрафторэтилиденовым фрагментом.
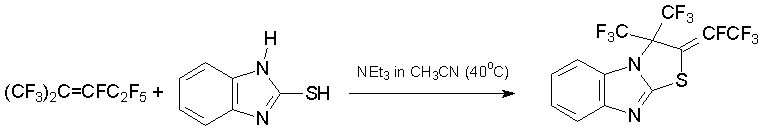
В то же время конденсация алкена 3 или азалкена 6 с 1N,3N-бинуклеофилами даёт шестичленные гетероциклы – производные пиримидина или 1,3,5-триазина, соответственно. Это продемонстрировано примерами получения замещённых пиримидинонов из 2-аминобензотиазола, 2-аминотиазола, 2-амино-5-хлорбензоксазола [37], мочевины, бензамидинов и гуанидина [39, 40].
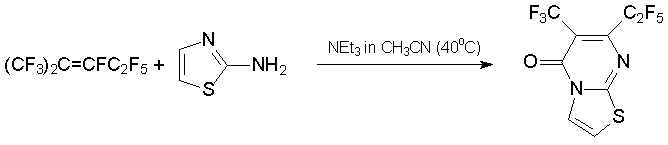
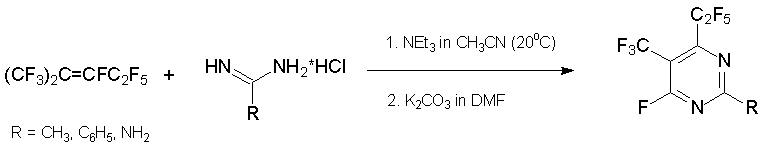
Родственные реакции с азалкеном 6 дают 2-R-4,6-бис(гептафторпропил)-1,3,5-триазин. Использование в качестве бинуклеофила мочевины приводит к замещённому 1-окситриазину, который был выделен в кето-форме [39, 40].
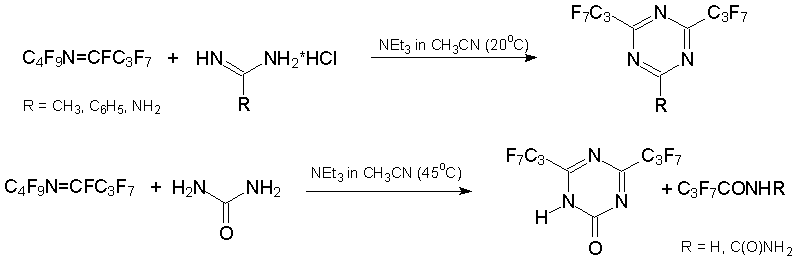
При взаимодействии с 1N,4N-, 1O,4O- и 1N,4O-бинуклеофилами формируются семичленные гетероциклические системы. Так, из азаалкена 6 и этиленгликоля или этилендиамина синтезированы производные дигидродиоксазепина и триазепина, соответственно, а в реакции с производными 2-аминоэтанола получаются бис(гептафторпропил)дигидрооксадиазепины [41].
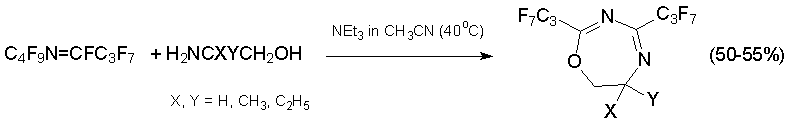
Из алкена 3 и производных 2-аминоэтанола получаются ди- и тетрагидрооксазепины, содержащие при С=С связи трифторметильную и пентафторэтильную группы. Ожидалось, что взаимодействие с этилендиамином даст соответствующий диазепин, но вместо него был выделен замещённый диазабициклононадиен, продукт дальнейшей реакции диазепина с алкеном 3 [41, 42].
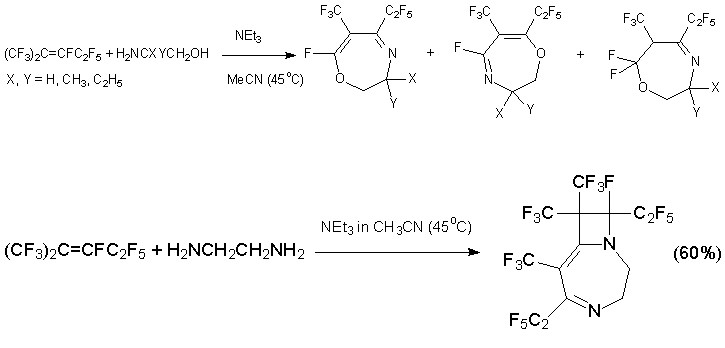
В тех же условиях из 1,6-гексаметилендиамина (1N,7N-бинуклеофил) получается диазабициклоалкен другого типа [42].
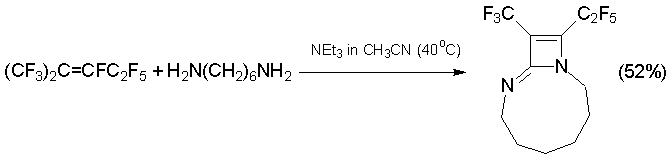
В представленных примерах описано получение гетероциклических соединений, начальной стадией которого является нуклеофильное замещение атома фтора во фрагменте =CF-C перфторалкена (перфторазаалакена). Однако возможен другой путь формирования гетероциклической системы – реакция нуклеофилов с полифторированными алкенами, содержащими фрагмент =C(X)-C, где Х – полифункциональный заместитель. Это было наглядно представлено синтезами производных тиазола и тиазина из перфтор-2-метил-3-изотиоцианат-2-пентена (7) и ряда O-, N-, S- и P-нуклеофилов.
Полифторалкен 7 синтезирован с высоким выходом реакцией перфторированного 2-метил-2-пентена 6 с роданидом натрия или калия (Раздел 2).
О-Нуклеофилы (спирты, фенолы) реагируют с алкеном 7 в отсутствии основания, образуя эфиры тиокарбаминовой кислоты (CF3)2CHC(C2F5)=NC(S)OR. Под действием основания (K2CO3 в ДМФА, 50-70°С) они циклизуются в производные 2-алкокситиазола и тиазолидинона. Эти гетероциклы получаются напрямую нагреванием алкена 7 с ROH в присутствии триэтиламина [43, 44].
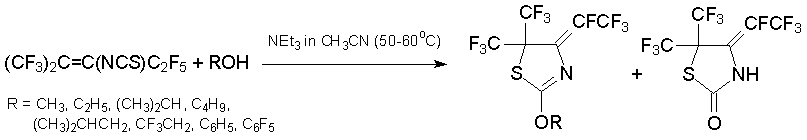
Реакции алкена 7 с N-нуклеофилами протекают неоднозначно. Так, в избытке метиламина с высоким выходом образуется замещённый 3Н-пиримидин-4-тион [45].
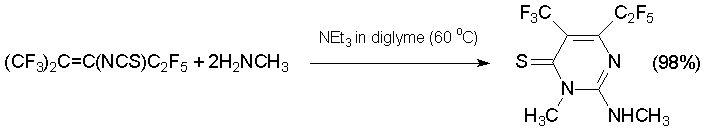
В реакции со вторичными аминами в мягких условиях циклизация приводит к шестичленным гетероциклам иного типа, 2-замещённым производным 5-трифторметил-4-пентафторэтил-6Н-1,3-тиазина [46, 47].
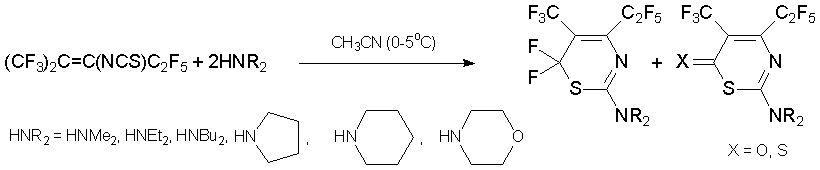
Взаимодействие с азолами в присутствии триэтиламина даёт с высокими выходами 2-замещённые производные 4,5-дигидротиазола [48, 49]. Примечателен факт, что амбидентные 1N,3O- и 1N,3S-бинуклеофилы реагируют с алкеном 7 только атомом азота [50].
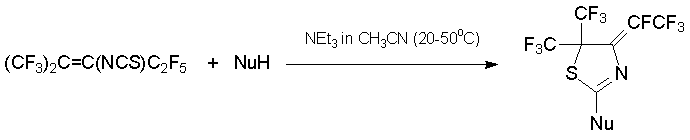
NuH = имидазол, пиразол, 1,2,4-триазол, бензотриазол, карбазол, фенотиазин, бензимидазол, 2-меркаптобензоксазол, 2-меркаптотиазол, 2-меркаптопиридин
Результат реакции монодентадных S-нуклеофилов зависит от основности среды и характера основания. В отсутствии такового алкен 7 под действием тиолов превращается в тиоэфиры N-(перфтор-2-метил-2-Н-пент-3-илиден)дитиокарбаминовой кислоты. Нагревание алкена 7 с бутантиолом и поташом в ДМФА приводит к смеси 3-бутилтиопентафтор-2-метил-2-пентена и замещённого дигидротиазола, а с триэтиламином в ацетонитриле образуются только производные дигидротиазола. Такой же результат получается в реакции с заведомыми тиолатами натрия или калия [51, 52].
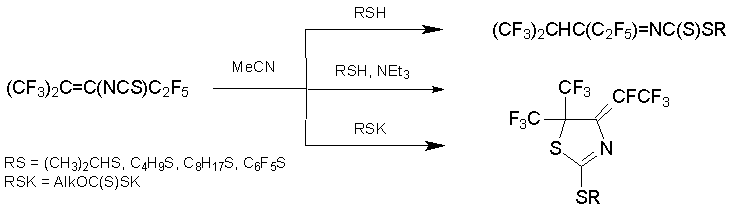
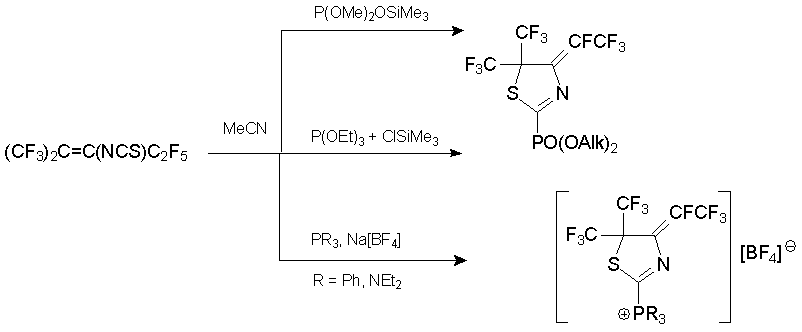
Ряд фосфорсодержащих дигидротиазолов получен из алкена 7 и P(III)-нуклеофилов. В присутствии веществ, связывающих фторид анион, с высокими выходами образуются производные тиазолилфосфорной кислоты или соли фосфония [53].
Список литературы
Материал рекомендован к публикации членом редколлегии В.Е. Платоновым
Fluorine Notes, 2012, 82, 1-2
