Received: April, 2011
Fluorine Notes, 2012, 81, 1-2
RADICAL REACTIONS OF POLYFLUOROARENES
L. S. Kobrina
N.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry of the Siberian Branch of Russian Academy of Sciences, Novosibirsk, Russia
E-mail: kobrina@nioch.nsc.ru
Abstract: Reactions of polyfluoroaromatic compounds with a wide range of radicals of different polarity and steric demands are discussed. The traditional notion on an intermediacy of (polyfluorinated in this case) cyclohexadienyl radicals (radical σ-complexes) can be applied to these reactions. The specificity of polyfluoroarenes as substrates manifest itself in the radical reactions both in the predominant dimerization of the intermediate polyfluorinated radical σ-complexes and in their isomerization via the first discovered fluorine 1,2-shift from an sp3-carbon to the near-by radical site.
Keywords: polyfluoroarenes, radical reactions, polyfluorinated cyclohexadienyl radicals, fluorine 1,2-shift.
The free radical attack on a substrate is one of the main types of interaction in organic chemistry, interest to which is caused by the fact that this mechanism is realised in a vide diversity of reactions which can be applied in synthesis and industry.
From the point of studying of the mechanism of radical reaction of the aromatic compounds polyfluoroaromatic compounds are a principal new type of objects allowing to study the influence of some effects absent in hydrocarbon systems on radical reactions mechanism. These are in the first place the possibility of radical attack on the aromatic ring position occupied by fluorine atom and consequently, the effect of fluorine atom as an ipso-substituent on the reaction rate and and as geminal substituent in intermediate radical σ-complexes on the route of their transformation. Secondly, this is the total effect of non-ipso fluorine atoms in an aromatic substrate (or in unsaturated part of intermediate) on the same reaction characteristics.
The systematic study of radical reactions of basic polyfluoroarenes – hexafluorobenzene, octafluorotoluene, decafluorobiphenyl and octafluoronaphthalene with the wide range of radicals of variable polarity and steric demands allowed to judge on the influence of these radical characteristics on the course and results of polyfluoroarene radical reactions and justify the possibility to spread the mechanistic ideas, which are conventional for the radical reactions of non-fluorinated arenes, on polyfluoroarenes [1, 2].
The Reactions of Polyfluorinated Benzene Derivatives
For the radical σ-complexes formed in the polyfluoroarene reactions with C-centered radicals three main ways of stabilization are typical depending on polar properties of the radical, the method of its generation, and reaction conditions: defluorination to yield products of homolytic substitution, dimerization, and recombination.
The interaction of hexafluorobenzene with methyl radical generated by thermolysis of di-tert-butyl peroxide [3] (140°C) results in the predominant formation of a mixture of the steric isomers of bis(methyl)perfluorotetrahydrobiphenyls 3a–5a. Defluorination of these σ-complex recombination products gave 4,4’-, 3,4’- and 3,3’-bis(methyl)octafluorobiphenyls 6a-8a in the quantitative yields. The structures of these products indicate that σ-complex (1a) initially formed by the addition of methyl radical (CH3•) to hexafluorobenzene underwent partially the rearrangement to σ-complex 2a by the fluorine 1,2-shift (Scheme 1) [3].
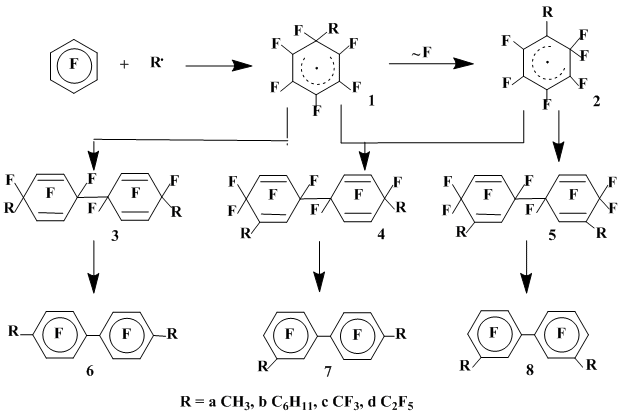
Scheme 1
Unlike this, with CH3• generated from acetyl peroxide at 80°C hexafluorobenzene gave a mixture consisting mainly of the steric isomers of tetrahydrodiphenyl derivative 3a, the products 4a and 5a of recombination of the isomerized σ-complexes being present in minor quantities [4, 5]. The latter is probably caused by the easiness of acetyl peroxide decomposition and, as a consequence, the occurrence of hexafluorobenzene interaction with CH3• at the relatively low temperature thus retarding the rearrangement of the initially formed σ-complex. As a different reason, one may consider the faster decomposition rate of acetyl peroxide compared to di-tert-butyl peroxide which obviously results in the higher σ-complex concentration and, as a consequence, in the shortening of its life time due to acceleration of the recombination. The distinction in transformations of the σ-complex formed by C6F6 with CH3•, originating from different sources, (aromatization in the reactions of C6F6 with CH3NO2 [6] at 550 °C and recombination in the reactions with acetyl and di-tert-butyl peroxides [3]) is probably caused by the different availability of radicals capable to effectively abstract a fluorine atom from the sp3 carbon of the radical σ-complex.
The homolytic fluorine substitution in polyfluoroarenes prevails when the system MeMgI–MHlgn (M = Ag or Cu) in diethyl ether is used as a source of CH3• (for the mechanism of the CH3• generation under these conditions see [7, 8, 9]), which is due to the presence in the reaction medium of agents (for example, reduced metals) capable to effectively aromatize the radical σ-complexes by removing the geminal fluorine atom. Along with methyl containing compounds, the products of fluorine substitution by secondary radical EtOCH•CH3, generated, obviously, by the attack of diethyl ether by CH3• [10, 11]:
CH3MgI
+ MX → CH3M
+ MgIX
CH3M → M + .CH3
.CH3 + CH3CH2OCH2CH3 → CH4 + CH3.CHOCH2CH3
The isomers distribution of the octafluorotoluene methylation products is o:m:p= 37:24:39. For substrates C6F5X (X = F, CF3, C6F5) the ratio of the competing fluorine substitutions by CH3• and by EtOCH•CH3 depends on the substituent X character: in going from X = F to X = CF3 and then to X = C6F5 the yield of methyl substituted products decreases in favour of 1-ethoxyethylsubstituted products, from decafluorobiphenyl practically only isomeric 1-ethoxyethylnonafluorobiphenyls being formed (Scheme 2) [12].
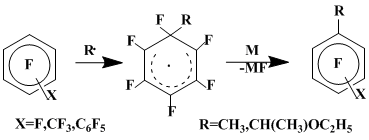
Scheme 2
The hexafluorobenzene interaction with the cyclohexyl radical (C6H11•) generated by thermolysis (140°C) of di-tret-butyl peroxide in cyclohexane proceeds mainly through recombination and dimerization of initially formed σ-complex 1b and its isomer 2b, emerging from 1b due to the fluorine 1,2-shift (Scheme 1). Like the above reactions of hexafluorobenzene with CH3•, generated by thermolysis of peroxides, the products of recombination of the σ-complexes derived by C6H11• are the mixtures of steric isomers, defluorination of which leads to isomeric dicyclohexyloctafluorobiphenyls 6b–8b [13].
However, the reaction of hexafluorobenzene with C6H11• in the presence of compounds capable aromatize the initially formed σ-complex (the dimer of pentafluorophenoxyl radical, molecular oxygen, di-tert-butyl peroxide) leads to the significant formation of cyclohexylpentafluorobenzene (20%) along with the product of the σ-complexes 3b–5b recombination. Obviously, the pentafluorophenoxyl radical (C6F5O•) is responsible for the aromatization, abstracting the fluorine atom from the radical σ-complex and yielding hexafluorocyclohexadienon. This is in line with the efficiency of pentafluorophenol additive to increase the polyfluoroarene phenylation [14] and with the propensity of pentafluorophenol to be easily transformed to C6F5O• under the reaction conditions [15] (Scheme 3).
Both primary σ-complexes and their through fluorine shift rearranged isomers can be easily aromatized. At that, the correlation of the σ-complex dimerization and recombination does not depend on whether the reaction is carried out in the inert atmosphere or under aerobic conditions. Obviously, this exhibits nearly the same defluorination rates of both non-isomerized and isomerized σ-complexes.
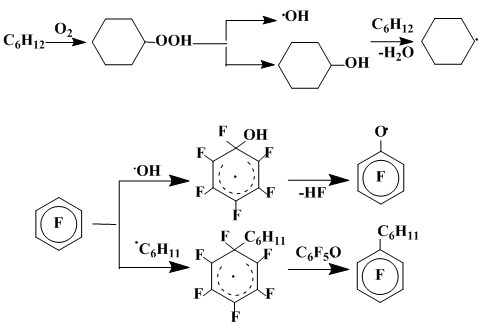
Scheme 3
The hexafluorobenzene interaction with trifluoromethyl (CF3•) and perfluoroethyl (C2F5•) radical, generated thermolytically (80oC) from perfluoroacetyl and perfluoropropanoyl peroxides, respectively [3, 4, 5], affords mainly the σ-complex dimers probably as mixtures of the steric isomers. Their defluorination yields quantitatively perfluoro-4-4’-bis(alkyl)biphenyl 6c,d (Scheme 1). The structures of polyfluorobiphenyls thus obtained proves that, as opposed to nonfluorinated alkyl radical, the σ-complexes derived by perfluoroalkyl radicals do not undergo the fluorine migration.
The interaction of decafluorobiphenyl with CF3• generated by thermolysis of trifluoroacetyl peroxide is rather complicated [16]. The mixture of perfluorophenyl(dimethyl)cyclohexadienes 9-11 (18%), which are the mixtures of geometrical isomers differing by the mutual disposition of trifluoromethyl groups, is formed along with the complex mixture of the σ-complexes dimers (13%) derived by the CF3• addition to decafluorobiphenyl. By defluorination of these dimers perfluorobis(phenyl)bis(methyl)biphenyls were quantitatively obtained. The formation of the above compounds means that the radical σ- complexes forming in the course of the reaction, undergo not only dimerization but also recombination with CF3• (Scheme 4).
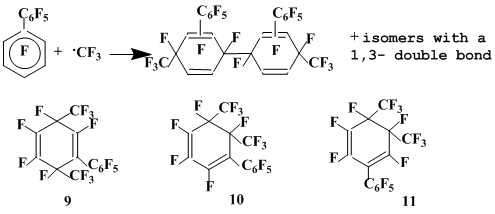
Scheme 4
As in the hexafluorobenzene reactions with CF3• и C2F5•, the σ-complexes formed by decafluorobiphenyl with CF3• do not rearrange via the fluorine 1,2-migration.
Depending on the reaction conditions [17], the hexafluorobenzene interaction with benzoyl peroxide results in formation of 6 to 50% of the dimers of σ-complexes derived by the C6H5• addition to hexafluorobenzene 12 and 13, defluorination of which produces quaterphenyls 16 and 17. Their structures reveals the rearrangement of initially formed σ-complex via the fluorine 1,2-migration (Table 1, Scheme 5).
Table 1. Interaction of benzoyl peroxide and hexafluorobenzene [17]
| Peroxide concentration | Т ºC | Time, hr | Yield % | 16 to 17 ratio | |
| C6H5C6F5 | C24H10F12 | ||||
0.0125 0.025 0.025 |
60 80 80 |
200 72 100 |
17 43.5 62.5 |
50 25 6 |
49:39 22:42 8:55 |
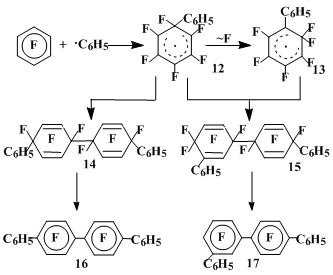
Scheme 5
In the hexafluorobenzene interaction with pentafluorobenzoyl hyperoxide practically only the recombination and dimerization of the radical σ-complexes are realized. At 80 °C the dimer 21 of complex 18 derived by pentafluorobenzoyloxyl radical (C6F5CO2•) is mainly (70% yield) formed. Besides, decafluorobiphenyl and perfluorophenylbenzoate (5% in total) as well as an insignificant amount of the mixture of the σ-complexes 22 and 23 recombination products were isolated (Scheme 6). These complexes contain both pentafluorobenzoyloxyl and pentafluorophenyl group [18].
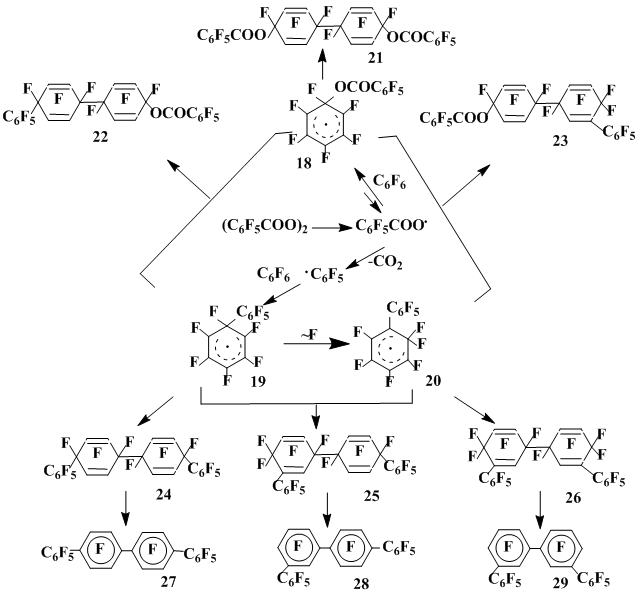
Scheme 6
Elevating the temperature (80–200°C), we achieved the formation of compounds corresponding to the pentafluorophenyl radical (C6F5•) addition to the C6F6 [19, 20]. Dimer 26 of complex (20) being a main product at 200 °C [19]. The structure of dimer 26 was established by by the 19F NMR and confirmed by the quantitative defluorination to m,m'-perfluoroquaterphenyl 29, thus proving that σ-complex 19 initially formed via the C6F5• addition to hexafluorobenzene undergoes the fluorine 1,2-shift. This first demonstrated fluorine migration from a sp3-carbon to the neighboring radical centre is the most important distinctive feature of the polyfluoroarene radical reactions.
Thus, within the temperature range of 80–200 °C (Table 2) we could obtain all possible products 21–26 of dimerization and recombination of radical σ-complexes 18–20 generated in the course of the hexafluorobenzene interaction with pentafluorobenzoyl peroxide [20] (unlike that the authors [21, 22] managed to identify only decafluorobiphenyl and perfluorophenylbenzoate in the 10% total yield of as the products of this reaction).
Considering the data listed in Table 2, one can see that elevating the temperature and decreasing the peroxide concentration enhances the yields of rearranged products 23, 25 and 26 and diminishes the yield of dimer 21. The increased rearrangement extent with lowering the peroxide concentration at the certain temperature can result from the longer radical σ-complex life time owing to its decreased concentration. At high peroxide concentrations the life period of unrearranged σ-complexes shortens, that leads to sharp decreasing of rearranged σ-complexes (look for dimers 23 and 26 content in Table 2).
Elevating the temperature and decreasing the peroxide concentration influence the ratio of the products formed from the σ-complexes derived by C6F5• and C6F5CO2• radicals in such a way that the yields of the first ones increase. This can be due to a reversibility of the C6F5CO2• addition to hexafluorobenzene and accelerating the radical decarboxylation. The study of the dimer 21 thermolysis in hexafluorobenzene and benzene at 200 °C gave a clear evidence of the reversibility of the C6F5CO2• addition [20].
Table 2. Decomposition of pentafluorobenzoyl peroxide in hexafluorobenzene [20]
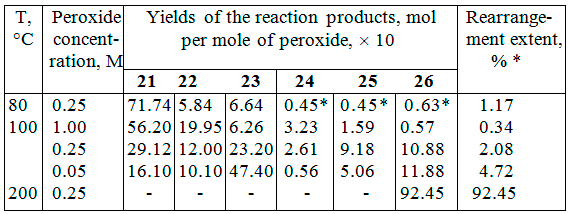
* calculated using the ratio (26 + 1/2 25) : (24 + 1/2 25)
The issue of reversibility or irreversibility of the C6F5• addition is one of the central subjects of the discussion of its interaction mechanism with polyfluoroarenes. The rationalization of pentafluorobenzoyl peroxide decomposition in hexafluorobenzene is based on the idea of irreversibility of this addition as well as of dimerization and recombination of all radical complexes forming in the process course at 80–100 °C. Such a notion is evoked by the fact that compounds 24–26 stay intact being kept in absolute benzene at 100 °C for 45 h [20].
Besides, incomplete thermolysis of dimer 24 at 200 °C exhibited that only the starting compound and dimer 26 are present in the reaction products [20]. The absence of compound 25 among the thermolysis products means that σ-complex 19 quickly undergoes the rearrangement into σ-complex 20 under these conditions. These results prove that the C6F5• addition to hexafluorobenzene is virtually irreversible till 200 °C. At 220 °C dimer 24 completely transforms into dimer 26 for 20 hr. Hexafluorobenzene was not found among the reaction products.
Dimer 26 remains intact within 20 h at 200 °C, which reveals the irreversibility of the whole process under these conditions proceeding consecutively via the C6F5• addition to hexafluorobenzene, rearrangement of the radical σ-complex thus formed and dimerization of the rearranged σ-complex. However, at 250 °C compound 26 transforms into decafluorobiphenyl [20].
The use of other potential sources of C6F5• to involve it into the interaction with hexafluorobenzene gave no such informative results. So, C6F5•. derived by photolysis of pentafluoroiodobenzene [23] or oxidation of pentafluorophenylhydrazin by silver oxide at 0 °C does not react with C6F6 [24]. However, at 100 °C the latter reaction results in the formation of above products 24-26 of hexafluorobenzene interaction with C6F5•, though their yields are not high, the main direction being the attack of starting C6F5NHNH2 by C6F5• to give pentafluorobenzene [25].
Besides, the mixture of two stereoisomeric perfluoro-4,4’-bis(phenyl)-1,1’-bicyclohexadienylidens 32 was isolated. Defluorination of this compound by zinc powder in glacial acetic acid results in the formation of p,p'-perfluoroquaterphenyl 27 in a high yield. The formation of compound 32 is obviously caused by the consequent addition of C6F5• to pentafluorophenylhydrazine, oxidation of thus derived σ-complex 30 by silver oxide resulting in the generation of dienocarbene 31, the dimerization of which completes the reaction (scheme 7). Generating of ketocarbene from quinondiazides can be an analogue for such a reaction scheme [26].
When using pentafluorophenylazotriphenylmethane as a source of C6F5• (80°C), the reaction of the latter with hexafluorobenzene led to a complex mixture containing traces of decafluorobiphenyl and dimer 26. Apparently, C6F5• thus formed recombines with triphenylmethyl radical, the stationary concentration of which is rather high, or interacts with the non-fluorinated moiety of pentafluorophenylazotriphenylmethane [3].
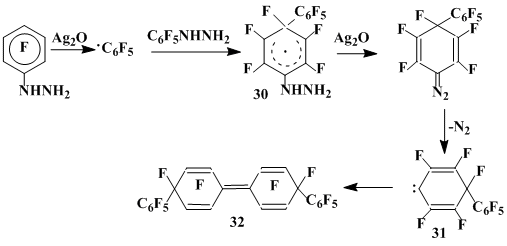
scheme 7
The study of the reactions of octafluorotoluene and decafluorobiphenyl with pentafluorobenzoyl peroxide allowed one to characterize the electronic and steric effects of trifluoromethyl and pentafluorophenyl groups on the regularities of radical reactions found out for hexafluorobenzene, particularly on relative ability of the radical σ-complexes to dimerize and rearrange via the fluorine migration. As opposed to hexafluorobenzene, octafluorotoluene [27] and decafluorobiphenyl [28] with pentafluorobenzoyl peroxide gave even under mild conditions (80 °C) the products of the isomerization and recombination of σ-complexes derived by the C6F5• addition mainly on the position meta to the non-fluorine substituent, along with small quantities of products corresponding to the same regioselectivity of homolytic substitution: perfluoromethylbiphenyls from octafluorotoluene and perfluoroterphenyls from decafluorobiphenyl (Scheme 8).
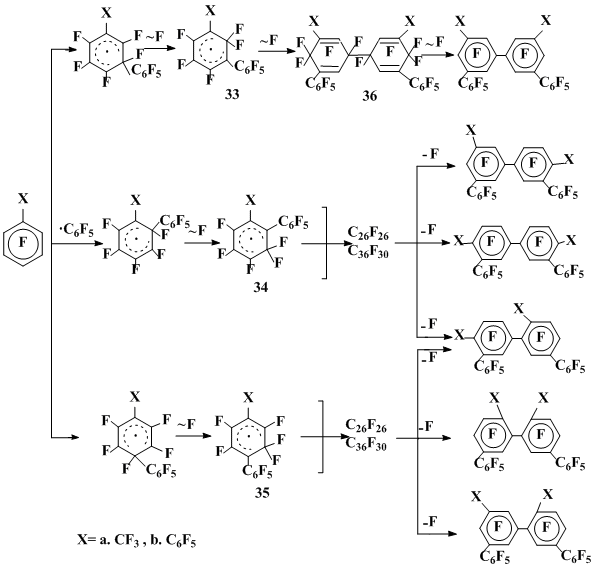
Scheme 8
Defluorination of dimers obtained from octafluorotoluene yields perfluoromethylbiphenyls along with perfluorobis(phenyl)bis(methyl)biphenyls, that may point out a partial dissociation of the dimers on the radical σ-complexes (Scheme 9).
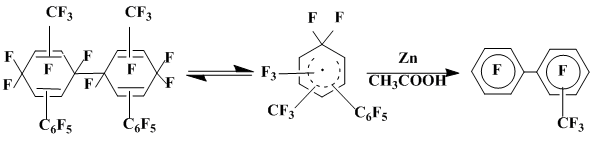
Scheme 9
The possibility of such dissociation is confirmed by the fact that in mass-spectra of the σ-complexes dimers the molecular ion is of small intensity or absent at all, but the most intensive is the ion with m/e = 1/2M.
Along with deriving the isomeric C26F26 products of σ-complexes recombination, the octafluorotoluene interaction with pentafluorobenzoyl peroxide also results in formation of noticeable amount of compounds C45F44 [27]. Judging from structure 38 of the isolated individual compound established by its defluorination results (compound 39), these products are derived from the interaction of dimer 36a and its isomers with C6F5• followed by the recombination of thus formed radical σ-complexes of type 37 with σ-complex 33a (Scheme 10).
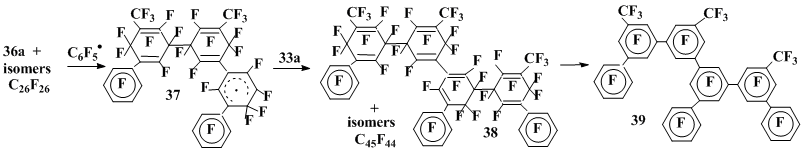
Scheme 10
Somewhat unexpectedly, the C26F26 dimers add C6F5• on the aromatic ring rather than on the double bond, despite polyfluorinated olefins are known to react with hydrocarbon radicals faster than polyfluoroarenes [29]. The lack of reaction on the double bond can be caused both by steric hindrances and smaller delocalization of the odd electron density in the respective reaction intermediate compared with that resulting from the attack on the aromatic ring.
As one would expect, due to the decreased substrate selectivity of a radical attack this side process is diminished under strong conditions (200 °C). Elevated temperature also influences the yield and isomer distribution of perfluoromethylbiphenyls formed by octafluorotoluene with pentafluorobenzoyl peroxide. Increasing the yields of these products with temperature probably testifies the bigger acceleration of the radical σ-complexes aromatization compared with their other transformations, whereas the nearly equal o-, m- and p-isomer distribution of the products of fluorine substitution obtained at 200 °C reflects decreasing the selectivity of the substrate attack by C6F5• with temperature. Data [30] on the reactions of non-fluorinated arenes with the phenyl radical allow one to believe that under these conditions the pentafluorophenyl migration does not occur.
The lack of products formed due to the substrate attack by C6F5CO2• in the octafluorotoluene and decafluorobiphenyl reactions with pentafluorobenzoyl peroxide is probably a consequence of smaller reactivity of these substrates towards the this electrophilic radical compared with hexafluorobenzene. Besides that, a certain role could be played by a lower stability of the σ-complexes formed by octafluorotoluene and decafluorobiphenyl with C6F5CO2• as well as their recombination products compared with those derived by hexafluorbenzene reversibility of their formation. In the aggregate with the practical irreversibility of C6F5CO2• decarboxylation this seems a possible reason of the observed result.
The regioselectivity found out for the octafluorotoluene and decafluorobiphenyl reactions with C6F5CO2• allows one to suggest that a relative stability of the isomeric intermediate radical σ-complexes is determined by a higher capability of fluorine to delocalize an odd electron compared with the trifluoromethyl and pentafluorophenyl groups. In the latter case this can be due to a noncoplanarity of pentafluorophenyl and cyclohexadienyl fragments. As a consequence, polyfluorinated σ-complexes are probably more stabilized by fluorine rather than by these substituents being located in the resonance positions of the cyclohexadienyl moiety.
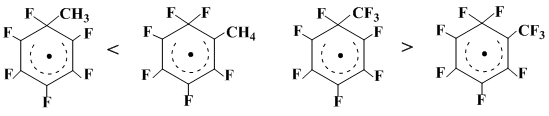
The above qualitative picture of the relative reactivity of hexafluorobenzene, octafluorotoluene and decafluorobiphenyl towards C6F5• is borne out (cf. the approach [31]) by a good compliance of the calculated and experimental relative reactivities of hexafluorobenzene and benzene in radical reactions and also by the well discernible dependency of the substrate reactivity on radical polar properties [32, 33]. The fruitfulness of this approach to characterize the reactivity of polyfluoroarenes toward free radicals is illustrated by the fact that with increasing electrophilicity in the sequence CH3• < C6H5• < CF3• < C6F5• the relative addition rate of these radicals to C6F6 drops compared with C6H6.
The hexafluorobenzene interaction with hydroxyl radical (HO•) generated by thermolysis (140 °C) of H2O2 in acetonitrile results in formation of pentafluorophenol, the further oxidation of which by the excess of H2O2 produces difluoromaleic acid. If pentafluorophenol is in the excess over H2O2, tetrakis(pentafluorophenoxy)benzochinon-1,4 is the principal product (Scheme 11) [34].
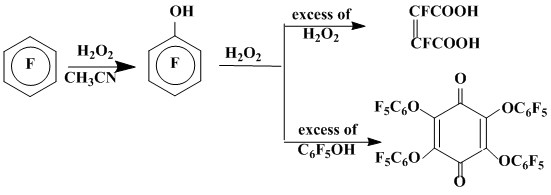
Scheme 11
Under analogous conditions mainly isomeric pentafluorophenylphenols are formed from pentafluorobiphenyl that reveals the preference of electrophilic HO• to add to unsubstituted aromatic ring.
Octafluoronaphthalene Reactions
The octafluoronaphthalene (OFN) interaction with different sources of CH3• results in fluorine substitution in the 1- and 2-positions and dimerization of the intermediate radical σ-complexes formed by the radical addition. Thus, thermolysis of di-tert-butyl peroxide in OFN (140 °C) in the inert atmosphere leads to a mixture of the above σ-complexes dimers 40 and 2-methylheptafluoronaphthalene 41 (in the 34% and 15% yields, respectively) [35]. At higher temperature (160 °C) an insignificant amount of 1-methylheptafluoronaphthalene 42 (5%) appears in addition to products 40 and 41, the yields of which increase (54% and 36%) (Scheme 12).
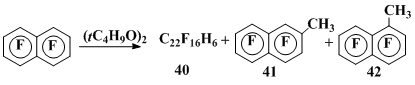
Scheme 12
The same interaction under aerobic conditions results in the exclusive formation of the fluorine substitution product – 2-methylheptafluoronaphthalene, i.e. defluorination becomes the main direction of the intermediate radical σ-complex transformation, similarly to what was discussed for the hexafluorobenzene reactions with alkyl radicals in the presence of dioxygen. When using methyl magnesiumiodide together with silver or copper halides in diethyl ether as a source of CH3•, OFN, like it was mentioned above for the benzene derivatives, undergoes mainly the fluorine substitution to produce the mixture of 1- and 2-methylheptafluoronaphthalenes in the 90:10 ratio. At that, along with methylheptafluoronaphthalenes the equal amounts of isomeric products of fluorine substitution by the secondary radical EtOCH2CH2• – 1- and 2-(α-ethoxy)ethylheptafluoro -naphthalenes – are also formed [12]. When using CuI, at the 40–55% OFN consumption, the 2:1 ratio of methyl- and (α-ethoxy)ethylheptafluoronaphthalenes does not change in the 1:(5–10) range of the CH3MgI/OFN ratio, and in the reaction with using AgCl it becomes 4:1 when the OFN concentration is increased twice.
Of the attention is also the fact, that the orientation of fluorine substitution in the OFN reactions with CH3• depends on the radical source: when using di-tert-butylperoxide 2-methylheptafluoronaphthalene is initially formed, and with CH3MgI-CuI(AgCl) – 1-methylheptafluoronaphthalene. The difference in temperatures (140 °C in the first case and 25 °C in the second) served as a ground to suppose that the regioselectivity alteration is caused by the reversibility of CH3• addition to OFN at high temperature and the irreversibility of the fluorine migration. The latter occurs only for the σ-complex derived by the CH3• addition on the OFN 2-position thus yielding 2-methylheptafluoronaphthalene if any defluorinating agents is present. Elevating the temperature to 160 °C accelerates defluorination thus yielding the minor amount of 1-methylheptafluoronaphthalene in addition to the 2-isomer (Scheme 13). Carrying out the reaction at the low temperature, i.e. on irreversibility of the CH3• addition, produces isomeric methylheptafluoronaphthalenes in the kinetically controlled ratio, the 1-isomer 42 being prevailing.
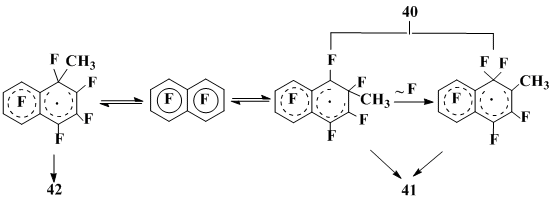
Scheme 13
The OFN interaction with benzoyl peroxide at 85 °C occurs as the fluorine substitution to yield 1-and 2-phenylheptafluoronaphthalenes in the 3.3:1 ratio and 1- and 2-benzoyloxyheptafluoronaphthalenes in the 1:1 ratio, along with perfluoro-2-(1’-naphthoxy)-1,4-naphthoquinone 46, hexafluoro-1,4-naphthoquinone 47 and products of the benzoyl peroxide transformation (biphenyl, phenylbenzoate, benzoylfluoride) [36]. In the acetonitrile medium the mixture is mainly formed of the products 45 of σ-complexes 43 and 44 dimerization and recombination (Scheme 14) [34].
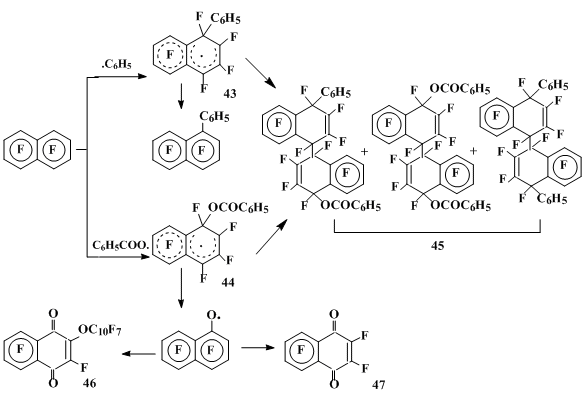
Scheme 14
The OFN reaction with pentafluorobenzenesulphonylfluoride as a source of C6F5• at 160 °C yields perfluoro-2-phenylnaphthalene (10%) (the insignificant amount of 1-isomer (2%) is formed only at 200 °C) and the mixture 50 (34%) of the recombination and dimerization products of both σ-complexes 48, derived by the pentafluorophenyl radical addition to the 2-position of OFN, and 49, arising from σ-complex 48 via the fluorine migration [35] (Scheme 15) [35].
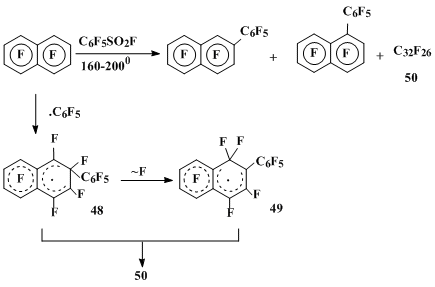
Scheme 15
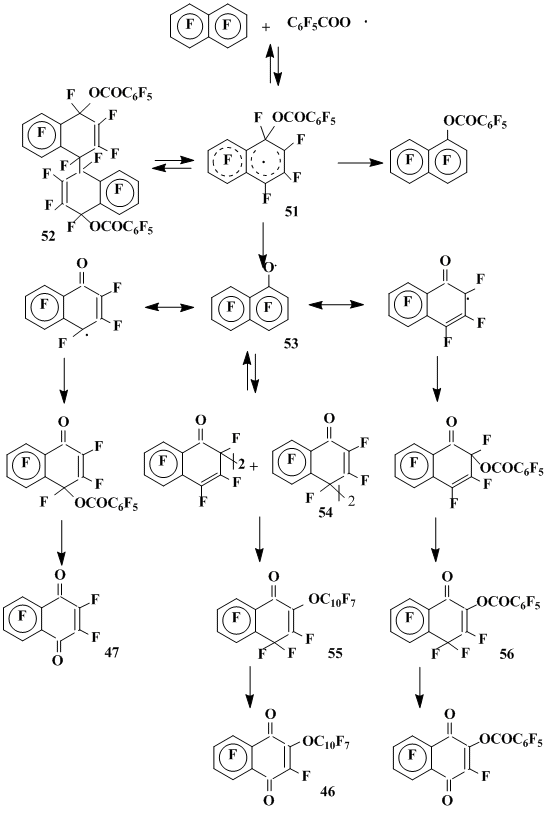
The OFN interaction with pentafluorobenzoyl peroxide in acetonitrile [37] at 80 °C results in the initial formation of the dimerization product 52 of σ-complex 51 derived by the pentafluorobenzyloxyl radical addition to the 1-position OFN. The process is obviously reversible as at 120 °C dimer 52 transforms into OFN and the mixture of compounds of the same composition as formed at 120 °C without a solvent. Perfluoro-1-oxo-2-(1’-naphthoxy)-1,4-dihydronaphthalene 55 and perfluoro-1-oxo-2-(benzoyloxy)-1,4-dihydronaphthalene 56 are the main products of the OFN interaction with pentafluorobenzoyl peroxide at 120°C without a solvent [38]. Besides, pentafluorobenzoylfluoride, hexafluoro-1,4-naphthoquinone 47 and the insignificant amount (1.5%) of the fluorine substitution products – 1- and 2-perfluoronaphtylbenzoates – are formed. The products derived by C6F5• were not found.
The formation of oxygen containing products in the OFN reactions with pentafluorobenzoyl peroxide has no a precedent among the known reactions of fluorinated compounds and, apparently, is the result of generating heptafluoronaphthoxyl radical in the reaction course. The isolation of pentafluorobenzoylfluoride from the products of this reaction is a serious argument in favor of the heptafluoronaphthoxyl radical 53 generation due to the fragmentation of σ-complex 51. The possible routes of the further radical 53 transformations are depicted in Scheme 16.
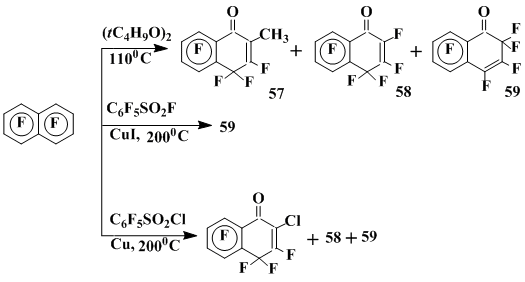
1-Oxoderivatives of polyfluoro-1,2- and -1,4-dihydronaphthalenes are the main products of the OFN reactions with di-tert-butyl peroxide at 110 °C and at high concentration of peroxide (0,25 M) or with pentafluorobenzenesulphonylhalides in the presence of copper and its salts. These products are formed by heptafluoronaphthoxyl radical, which is generated via fragmentation of the σ-complexes derived by OFN with tert-butoxyl or pentafluorophenylsulphonyl radical [35] (Scheme 17).
Scheme 16
Scheme 17
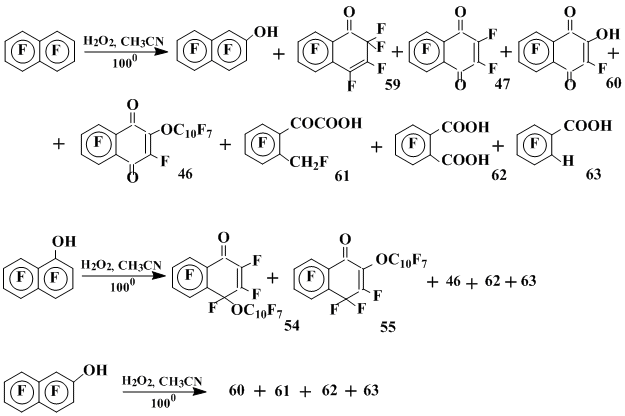
The products distribution derived by thermolysis of hydrogen peroxide in OFN (100°C) and also in heptafluoro-1- and -2-naphthol indirectly confirms heptafluoronaphthoxyl radical to form the oxygen containing products in the OFN reactions [39] (Scheme 18).
Scheme 18
The main products of these reactions are shown in Scheme 18: heptafluoro-2-naphthol, 1-oxoperfluoro-1,2-dihydronaphthalene 59, hexafluoro-1,4-naphthoquinone 47, 2-hydroxy-pentafluoro-1,4-naphthoquinone 60, perfluoro-2-(1’-naphthoxy)-1,4-naphthoquinone 57 and 2,3,4,5-tetrafluoro-6-(fluoromethyl)phenylglyoxylic acid 61. With a large excess of hydrogen peroxide the deep oxidation takes place to give tetrafluorophtalic 62 and 2,3,4,5-tetrafluorobenzoic 63 acids along with compounds 47, 57, 59 and 60 (Scheme 18).
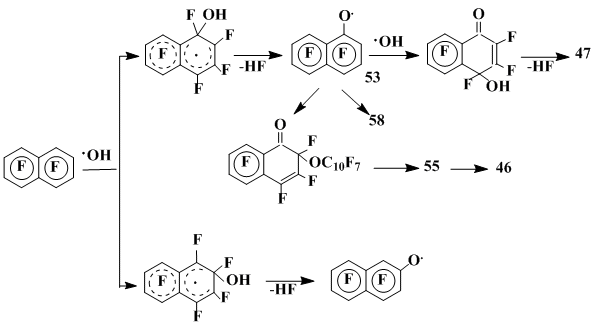
The examples of most important transformations of heptafluoronaphthoxyl radicals are given in Scheme 19.
Scheme 19
Transformation routes of polyfluorinated radical σ-complexes
The following transformations are principal for the radical σ-complexes formed in the course of reactions under consideration characterized by a generality of the first stage — ipso-addition of radical R• on the substrate position occupied by fluorine (Scheme 20):
- aromatization via the radical R• detachment and returning to the starting substrate;
- aromatization via the fluorine detachment to form a substitution product;
- fragmentation via elimination of a diamagnetic molecule from the geminal unit to form new polyfluorinated radical;
- recombination, including dimerization;
- rearrangement via the geminal fluorine 1,2-migration to the neighboring radical, which is unknown before for fluorine containing radicals.
As for the first stage of polyfluoroarene radical reactions, of the paramount importance are factors determining the rate of the radical addition to a substrate to form a radical σ-complex, the possibility of its return to the starting compound, and also the correlation of rates of these and other σ-complex transformations. From the attacking radical, the main role is played by its polarity, and from the substrate – by the number and mutual location of fluorine atoms.
Different types of competitive experiments to reveal the correlation of hexafluorobenzene and benzene reactivity confirmed the important role of polar properties of the attacking radical. Thus, electrophilic radicals add to benzene at a higher rate than to hexafluorobenzene, while the opposite takes place with nucleophilic radicals. For example, the ratios of addition rates to C6F6 and C6H6 are 0.44 for C6H5• at 80°C [40], 7.8 for CH3• at 65°C [29], 0.4 for CF3• and 0.8 for CH3CF2• at 50 °C [29].
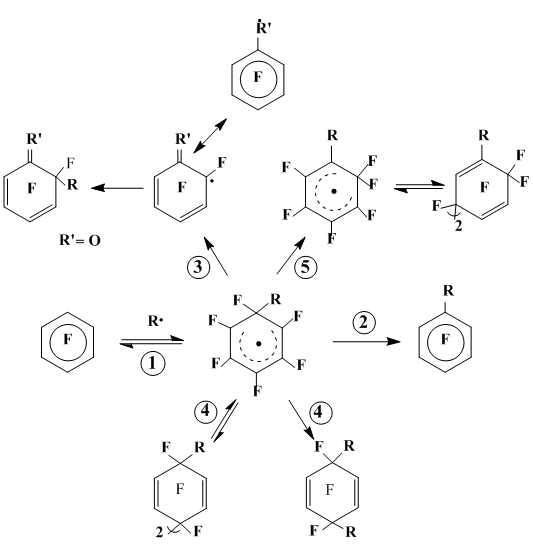
Scheme 20
In spite, that the key role of cyclohexadienyl radicals as intermediates in the radical reactions of arenes was suggested quite long ago, this notion was justified only indirectly, namely by the formation of these σ-complexes dimerization and also by the results obtained using the spin trapping method. Detection and direct observation of these intermediates was for a long time limited by specific cases, when they are highly stable due to steric reasons, associated, for example, with the presence of several tert-butyl [41] or trimethylsilyloxyl groups [42], and when they are formed by the OH• addition to the aromatic nucleus [43–45]. The ESR spectra of cyclohexadienyl radicals formed by the addition of Ph3Si• to benzonitrile [46], silyl, phosphonyl and methyl radicals to benzene [47] were registered as well.
The aggregate of these approaches was applied to prove the formation of cyclohexadienyl intermediates of polyfluoroarene radical reactions, that renders the validity of notions of the mechanism of these reactions not lesser than for non-fluorinated arenes.
The spin trapping method proved itself to be successful in studying the mechanism of homolytic reactions and establishing the nature of short-living radicals formed in the course of these reactions [48–53]. The respective data on polyfluoroarenes are sparse. However, they allow one to judge on the nature of radicals generated by thermolysis of the fluorine containing benzoylperoxides or phenylazotriphenylmethanes in the medium of benzene or hexafluorobenzene and also cyclohexadienyl radicals formed by these radicals addition to the substrates by using C-phenyl-N-tert-butylnitron (PBN) and nitrosodurene (ND) as spin traps,.
The ESR data [54] prove that, regardless of the number of fluorine atoms in aroyloxyl radicals formed by decomposition of fluorinated aroyl peroxides, their spin adducts of with PBN are characterized by the ESR parameters which are close to those for the PBN adducts with benzoyloxyl radicals [48–50] (αN =12.7–13.1, αo-H = 1.2–1.45 G).
The spin PBN adducts with fluorinated aryl radicals, the ESR spectra of which are analogous to those of the PBN adducts with substituted phenyl radicals [49, 51] (αN = 13.9–14.4, αo-H = 2.1–4.4 G), were registered only in cases of the decomposition of low-fluorinated aroyl peroxides. Analogously, the spin adducts of nitrosodurene were registered only with p-fluorophenyl radical generated by thermolysis of the corresponding peroxide [54]. At the same time, the respective spin adducts were reported to be registered upon generation of C6F5• from pentafluorophenylazotriphenylmethane in aromatic substrates in the presence of PBN [51] or ND [52].
The most basic and important is fixing the spin adducts of arylcyclohexadienyl radicals –intermediates of the radical reactions of arenes. When using PBN as a spin trap, we managed to register its spin adduct with pentafluorophenylcyclohexadienyl radical formed by benzene with C6F5• generated both by thermolysis of pentafluorobenzoyl peroxide [54] and pentafluorophenylazotriphenylmethane [51]. Upon thermolysis of fluorinated benzoyl peroxide (besides the above p-fluorobenzoyl peroxide) in benzene in the presence of ND at 100–115 °C the spin adducts are formed, displaying the ESR spectra in which STI with fluorine is absent and the STI constants αN, αo-H and αм-H are close to those for spin adducts of p-substituted phenyl radical with ND. Based on that, the registered radicals were suggested to be the spin adducts of p-arylsubstituted phenyl radicals and ND, but derived by oxidation of the initially formed spin adducts of arylcyclohexadienyl radicals [54] (Scheme 21).
Adduct 65a (X = H), along with its precursor – the spin adduct of pentafluorophenylcyclohexadienyl radical 64a (X = H), (αN = 13.2, αβ-H = 9.3 G), was registered upon generating C6F5• from pentafluorophenylazotriphenylmethane in benzene in the presence of ND [52].
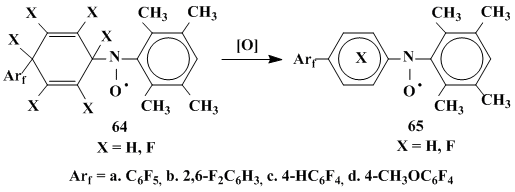
Scheme 21
For the cases of polyfluorene attack by the aryl radical, generated from an aroyl peroxide containing 4 or 5 fluorine atoms, the oxidation of the initially formed arylhexafluorocyclohexadienyl radical is significantly hampered. This allowed to register the spin adducts formed by polyfluoroarylhexafluorocyclocyclohexadienyl radicals with ND 64a,c,d X = F [54].
Thus, the application of the spin trapping method to study the reactions of hexafluorobenzene with fluorine containing aryl and aroyloxyl radicals provides an opportunity to register not only radicals attacking the substrate as the corresponding spin adducts, but also the intermediate polyfluorinated cyclohexadienyl radicals, that is of a principal importance to justify the ideas of the mechanism of polyfluoroarene radical reactions.
The use of the ESR method for direct observation and identification of cyclohexadienyl radicals formed in the polyfluoroarene reactions under study has been considered in the course of the discussion of rearrangement of polyfluorinated cyclohexadienyl radicals via the fluorine 1,2-migration we discovered.
The most typical transformation routes of polyfluorinated cyclohexadienyl radicals occurring in the course of polyfluorarene radical reactions are shown in Scheme 20. Only two of them are analogous to what was shown for ipso-reactions of haloarenes: the reversible addition of an attacking radical and aromatization of the cyclohexadienyl intermediate via the halogen removal.
The issue of the reversibility of the radical addition to a substrate is significant for the rationalization of orientation in the radical reactions of polyfluoroarenes. There are experimental reasons to consider that the addition of alkyl radicals is virtually irreversible within the wide temperature range (25–140 °C for CH3• and 140–200°C for cyclo-C6H11•) [13]. The addition of CF3• to hexafluorobenzene becomes reversible only above 180 °C [55]. There are no respective data on C2F5•, however, comparing the temperatures, at which the addition of CF3• and C2F5• to benzene becomes reversible (150 °C for CF3• [56, 57] and 110°C for C2F5• [58]), one may suppose that formation of the σ-complex of hexafluorobenzene with C2F5• will be reversible at lower temperature compared with CF3• .
The significant difference was found out in the temperature dependences of the difluorochloroethyl radical (generated by photolysis of 1,3-dichlorotetrafluoroaceton) addition rates to benzene and hexafluorobenzene which is due to the difference between temperatures at which the reversibility of these reactions becomes substantial [59]: for benzene – at 100°C, for hexafluorobenzene - at 250°C.
In spite that the above information is limited, one may sum up that the alkyl radicals addition to hexafluorobenzene becomes reversible at higher temperatures than to benzene.
Such a tendency can be expected to be kept for the addition of aryl radicals to arenes as well. Despite the fact that the data regarding the presence of isotope effect in these reactions are somewhat inconsistent, the addition of C6H5• generated by various methods [30, 60–66] to benzene is considered reversible only at about 200 °C. At the same time the addition of C6F5• to hexafluorobenzene is irreversible within 200–250 °C [20, 27, 28]. The addition of CH3• and C6F5• to OFN is reversible at lower temperature (140–160°C for CH3• and 200°C for C6F5• [35]) compared with hexafluorobenzene, that significantly determines the orientation in the case of OFN.
In the series of heteroatomic radicals involved in the reactions with polyfluoroarenes, only the addition of pentafluorobenzoyloxyl radical [19, 20, 27, 28, 30, 35, 38, 63–69] was strictly proved to be reversible. Basing on these data one may suppose that the main factor influencing the possibility of abstraction of the geminal fragment originating from the added radical is obviously the temperature rather than the nature of this radical.
The geminal fluorine detachment from a polyfluorinated σ-complex is one of the ways to a final product. In this case, either radical σ-complexes themselves or other components of a reaction mixture (radicals, metals and their halides, reaction vessel surface, air oxygen and others) can serve as halogen acceptors. Accordingly, in the reactions with nonfluorinated alkyl radicals, the opportunity of intermediate radical σ-complexes to undergo aromatization into a product of fluorine substitution depends on the method of radical generation, determining in turn the presence of different components. For example, on generating CH3• from acetyl peroxide (80°C) the yield of fluorine substitution product in hexafluorobenzene by methyl group doesn’t exceed 15–20%, the recombination of the σ-complex being the main reaction route [4]. At the same time, intermediate polyfluorinated cyclohexadienyl radicals undergo practically only defluorination in the reactions of polyfluorinated benzenes and OFN with alkyl radicals (CH3• and CHCH3OC2H5•) generated in the system CH3MgI–copper halide in diethyl ether. In these reactions, copper halide and reduced metal act obviously as effective defluorinating agents [12, 10].
A more effective defluorination of radical σ-complexes compared with thermal reactions is being observed when alkyl radicals are photolytically generated. Thus, upon the thermal generation of a cyclohexyl radical, even with air oxygen in a reaction vessel, the share of fluorine substitution in hexafluorobenzene does not exceed 20%, the σ-complex recombination being the main reaction direction under anaerobic conditions [13]. Aromatization of the σ-complex formed via addition of the photolitically generated cyclohexyl radical to hexafluorobenzene is predominant to afford cyclohexylpentafluorobenzene in 61% yield [70].
The contribution of defluorination of cyclohexadienyl radicals formed by polyfluoroarenes with non-fluorinated aryl radicals generated from aroyl peroxides is within the range from 30 to 80% for fluorinated benzenes [17, 71] and from 15 to 45% – for OFN [36]. Benzoic acid was suggested to be an effective defluorinating agent in this reaction. The formation of SiF4 in the reaction of OFN with benzoyl peroxide as a source of phenyl radical confirms that the reaction vessel wall may also be a participant of the radical σ-complexes defluorination [38, 36].
In most interactions of polyfluoroarenes with polyfluorinated alkyl and aryl radicals homolytic fluorine substitution, i.e. the above aromatization of a radical σ-complex, doesn’t occur. The fluorine gemination with a strongly electronegative polyfluorinated group renders this σ-complex resistant against defluorination. The strengthening of a C–F bond with accumulation of geminal fluorine atoms is known [72]. The higher stability of polyfluorinated arenium ions with two geminal fluorine atoms compared to isomers containing different group as a germinal substituent was also reported [73]. The reaction of OFN with pentafluorobenzene sulfofluoride serving for a source of pentafluorophenyl radical (200°C) is an exception, but the yield of perfluorophenylnaphthalenes doesn’t exceed 20% [35].
During the interaction of polyfluoroarenes with heteroatomic radicals the contribution of defluorination of intermediate radical σ-complexes in the total reaction also changes within a wide range. Thus, it is only about 2–10% for the interaction of hexafluorobenzene or OFN with benzoyloxyl [17, 36, 71] and pentafluorobenzoyloxyl [18, 20, 38] radicals generated thermally from corresponding aroyl peroxides, that undoubtedly is due to the small concentration of these radicals, the formation and transformations of which compete with other processes, such as generation of aryl radicals and their reactions with polyfluoroarenes. Homolytic substitution (50–60%) becomes predominant for the hexafluorobenzene interaction with photochemically generated silicon containing radicals (trimethyl- and trichlorosilyl) which themselves act as defluorinating agents [74].
The σ-complex fragmentation to generate new radical is actually the aromatization with the fluorine detaching but when the fragment of a substituent geminal to it serves as a fluorine acceptor.
Apparently, carrying out such a process requires the respective easiness of a X–Y bond cleavage and, possibly, an increased strength of the Y–F bond. The fragmentation of such type takes place, for example, during the interaction of OFN with oxygen containing radicals [35, 36, 38, 39] (Scheme 22).
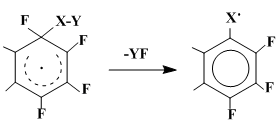
Scheme 22
When XY = ArCOO, a fluoroanhydride of the corresponding benzoic acid had been isolated as a product of the YF fragmentation. The another product of the radical σ-complex fragmentation – heptafluoronaphthoxyl radical – turns further into oxoderivatives of polyfluorinated dihydronaphthalenes and polyfluoronaphthoquinones, which are the final products of these reactions (Scheme 16).
It can be supposed, that the fragmentation to produce a new radical is the main type of stabilization of the σ-complexes formed by hexafluorobenzene [6] with O2N• or HO• [41,75]. The elimination of NOF in the first case and HF in the second one must give a pentafluorophenoxyl radical. Accordingly, pentafluorophenol was isolated in both cases, and dimer of pentafluorophenoxyl radical was registered using UV-spectroscopy in the course of hexafluorobenzene interaction with HO• [75].
Recombination, including dimerization, is the most typical transformation of polyfluorinated cyclohexadienyl radicals. The recombination with the initially generated radical to yield polyfluorinated cyclohexadien is a main way to stabilize the intermediate σ-complex in the reaction of decafluorobiphenyl with CF3•. Comparison of these data with the results of the hexafluorobenzene interaction with CF3•, which leads to the dimers of the respective intermediate σ-complexes, allowed one to relate the possibility of dimerization of the σ-complexes formed by C12F10 with CF3• to the rather long life of cyclohexadienyl radical in the cell of highly viscous decafluorobiphenyl used as a reaction medium [16] ( [29]).
The recombination is a main direction of the radical σ-complexes stabilization in the cases when aromatization via the fluorine atom abstraction is hampered by the absence of effective defluorinating agents. It is also evident, that due to the high C–F bond strength the recombination appears more preferable, than disproportion, which as well as the recombination requires two σ-complexes to encounter. The products of recombination and dimerization of polyfluorinated radical σ-complexes are, as a rule, formed in high yields virtually regardless of temperature and concentration of the radical source. For example, in the reaction of C6F6 with (C6F5COO)2 the total yields of these products reach 90–98% within the temperature range of 80 to 200°C and the peroxide concentration range of 0.25 to 1 M [20]. Of the exception are the cases when the above factors provoke the competitive processes. Thus, during decomposition of perfluoropropionyl peroxide in hexafluorobenzene [5] the increase in reaction temperature from 50 to 80°C results in almost double decrease in the yield of dimerization products of the hexafluorobenzene σ-complex with C2F5•, and the 25-fold increase in the peroxide concentration (the peroxide to hexafluorobenzene ratio is 1 : 40 and 1 : 1.5, respectively) decreases the dimers yield a ten times. In the reaction of hexafluorobenzene with acetyle peroxide [4] the analogous increase of peroxide concentration leads to the practically full disappearance of the corresponding σ-complexes dimers from the reaction products [3].
There are also indications that the recombination of σ-complexes under the conditions of these reactions is practically irreversible. For example, a continuous keeping the σ-complexes recombination products with the radical source under the corresponding reaction conditions neither brings about their mutual transformations, nor yields any other products [3]. The currently known cases of homolysis of the recombination or dimerization products to the corresponding σ-complexes occurs as a rule at the much higher temperatures compared with their formation through the radical attack on a polyfluoroarene.
The rearrangement by the fluorine 1,2-shift from a saturated carbon atom to the near-by radical centre is one of most important stabilizing ways of polyfluorinated radical σ-complexes. As the first example of a free radical isomerization of this type [19], by its significance this transformation goes beyond the polyfluoroarene chemistry and relates to the general problem of radical rearrangements through the halogen migration, which even in the middle of the last century were considered impossible (see, for example, [76]). In 1962 the first review was published, in which the information was summed up on the 1,2-shifts of hydrogen and chlorine atoms, aryl and alkyl groups in free radicals occuring on solutions [77]. In succeeding years the experimental data regarding radical rearrangements had been constantly accumulated, and virtually all reviews dedicated to radical reactions in general include the consideration of free radical rearrangements [78–81].
This literature proves that polyhaloalkyl radical rearrangements caused by chlorine migration were mainly known till 1980 [77, 81, 82]. Experimental material accumulated by now witnesses that 1,2-migration of chlorine atom occurs intramolecularly [77], and the results of quantum calculations show [83, 84] that it goes through a bridge-like transition state with a relatively low energy barrier (26.4 kcal/mol) [84]. It should be noted, that in all of cases, when chlorine and fluorine atoms compete, a chlorine atom is being shifted [85, 86]. The bromine migration in the polyhaloradicals proceeds at higher rate than the chlorine migration (1010 and 108 s-1, respectively), the latter being therefore observed more seldom [87].
The bromine 1,2-migrations in cyclohexadienyl radicals are most illustrative. Thus, analysis of the ESR spectra reveals that the cyclohexadienyl radicals with a bromine in the geminal position generated by the X-ray irradiation of methanol solutions of bromobenzene and para-bromophenol at 77 C° undergo the fast bromine 1,2-shift (the life time of the registered state is 10–7 sec) [88].
As mentioned above, the fluorine 1,2-migration in radicals was first discovered [19] and studied by us [3] for polyfluorinated cyclohexadienyl radicals. However, before starting to analyze the peculiarities of these transformations, it sees sense reasonable to briefly consider the later information regarding rearrangements of more simple fluorine containing radicals including the fluorine 1,2-migration. Thus, a fluorine migration in the 1,2-difluoroethyl radical generated by the tritium addition to trans- or cis-1,2-difluoroethylene was discovered [89]:
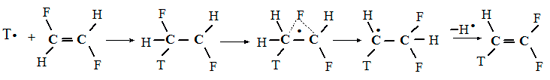
Japanese chemists [90] explained the formation of CF2=CHТ from the tritium action on 1,1,2-trifluoroethylene by the fluorine 1,2-migration in the initially formed ĊF2CHТF radical, believing that the difference in the C–F and C–H bond strength is not a reason to consider that the direct intermolecular transfer is possible. The latter requires preferable fluorine abstraction from the above radical. Basing on this, to explain the unexpected high yield formation of CF3CH2CH2CF3 as a result of hydrogen atom addition to trifluoroethylene [91] the Japanese researchers [92] suggested the fluorine 1,2-migration in the initially formed CF2CH2F• radical followed by the dimerization of thus derived radical CF3CH2•. Also these authors measured the fluorine atom migration rate constant as 2·1010 s–1.
It is known that the hydrogen 1,2-migration in the ethyl radical does not occur [80], and the energy of the C–F bond in fluorinated hydrocarbons is somewhat higher compared with the C–H bond. This obvious mismatch provoked a theoretical explanation of the preference of fluorine intramolecular migration. The molecular orbital calculations revealed the high energy barriers for the hydrogen and fluorine 1,2-shift [83, 93–96]. In the work [97] the stabilization of β-haloethyl radicals due to forming a halogen bridge was evaluated within the perturbation theory:
–∆E (kcal/mole): I (7.54), Br (2.97), Cl (0.22), F (–1,30).
These data argue for the fluorine bridge-forming to be unfavorable. According to the author’s [83] point of view, the reason for that is a large energy difference between the interacting orbitals which decreases in going from fluorine to chlorine, bromine and iodine. The halogen readiness to the 1,2-migration increases in the same direction.
On the other hand, the INDO calculations testify that the energy barrier of the fluorine 1,2-migration in 1,2-difluoro-, 1,1,2-trifluoro- [53] and 1,1,2,2-tetrafluoroethyl radical [98] via a transient bridge-like structure is twice lower compared with hydrogen.
Depending on the calculation method, the energy barrier of the fluorine 1,2-migration in 2-fluoroethyl radical varies from 28.6 to 107 kcal/mole [83,96], that elucidates certain difficulties in the theoretical interpretation of such migration. In the review [99] the radical rearrangements caused by the migration of fluorine and other halogens are considered more comprehensively.
The fluorine 1,2-shift in polyfluorinated cyclohexadienyl radicals was borne out by means of analysis of the kinetics of the hexafluorobenzene reaction with cyclohexyl radical [13] is monomolecular process. The yield ratio of the dimerization products of rearranged (R) and non-rearranged (NR) complexes is a linear function of radical concentration, the latter depending on the initial concentration of the radical source (RS):
NR/R = K[R•] = f [RS]0.
By the example of the hexafluorobenzene interactions with phenyl, pentafluorophenyl and cyclohexyl radicals it had been established that elevating temperature and decreasing the RS (radical source) concentration enhance the rearrangement degree [3] (see, for example, Table 2).
For the reaction of hexafluorobenzene with methyl and cyclohexyl radicals, which are close in polarity but appreciably differing in bulkiness, a steric hindrance associated with the sp3-unit in polyfluorinated radical σ-complex was shown to insignificantly influence a possibility of the fluorine 1,2-migration. This is illustrated (Table 3) by the alteration of the rearrangement degree (the yield ratio of the R and NR σ-complexe dimers formed by hexafluorobenzene with the respective radicals) as dependent on the RS concentration [3].
Table 3. The rearrangement degree in the reaction of hexafluorbenzene with CH3 (generated from di-tert-butyl peroxide) and C6H11• (generated from cyclohexane and di-tert-butyl peroxide, 140°C in both cases) [3].
[(t-BuO)2]0, M |
Radical |
|
CH3• |
C6H11• |
|
0.77 0.42 0.38 0.33 0.19 |
0.29 0.50 0.58 0.85 1.12 |
0.27 0.32 0.47 0.58 0.68 |
To estimate the influence of the geminal substituent nature in a polyfluorinated radical σ-complex on the possibility of the fluorine 1,2-shift, full energies and spin density distributions of the σ-complexes of C6F6 with CH3• or CF3•, on one hand, and the respective rearranged σ-complexes, on the other, were calculated by L.N. Schegoleva with using the INDO UHF method with the standard Pople-Segal parametriazation [3]. The calculation results show that the fluorine 1,2-shift in the σ-complex of C6F6 with CH3 leads to its 11,6 kcal/mol more stable isomer, whereas the situation is vice versa for CF3•: the initially formed σ-complex with the ring –FCCF3– unit is more stable by 3.8 kcal/mol compared with the rearranged one (Scheme 23).
Scheme 23
The recent theoretical study showed that the energy barrier (ΔE#) of the fluorine 1,2-shift in fluorinated cyclohexadienyl radicals is significantly lower than in alkyl radicals and the rearrangement occurs likely via a bridge-like transition state [100].
Thus, a driving force of the fluorine 1,2-migration can be an energy gain accompanying the formation of the –CF2– group due to strengthening the C–F bond with the accumulation of germinal fluorine atoms [84]. However, polyfluorinated radical σ-complexes with geminal fluorine and a highly electrononegative group (perfluoroalkyl or pentafluorobenzoyloxyl) are not prone to such a rearrangement.
This is confirmed by the direct ESR observations of polyfluorinated cyclohexadienyl radicals formed by in homolytical reactions of polyfluoroarenes [101]. First such observations were made for the radicals derived by the addition of protium and deuterium atoms to hexafluorobenzene and generated by the X-ray irradiation of C6F6 in adamantine and adamant-d16-ine matrix, respectively [102]. The ESR detection of hexafluorocyclohexadienyl radicals in a solution proved to be technically more simple compared with the respective nonfluorinated analogues by the higher signal intensity reason. This allowed one to obtain ample data on the structure of polyfluorinated cyclohexadienyl radicals and the rate constants of their formation by the radicals addition to hexafluorobenzene [103]. Thus, registered were polyfluorinated cyclohexadienyl radicals (RC6F6•) generated by photolysis of the radical sources in C6F6 at 25 °C. The HFI constants for thus generated and registered HC6F6• are in compliance with the analogous characteristics of the same radical generated in an adamantine matrix [102].
The added radical R• nature manifests itself in the ESR spectra of the cyclohexadienyl radicals derived from C6F6 in the HFI constant value of the ipso-fluorine atom, which changes from 65 G for R• = C2H5O• till 152 G for R• = (C2H5)3Si•. These values reflect steric and electron properties of R• and also provide the information on the structure of hexafluorocyclohexadienyl fragment. Thus, comparing the F1 HFI constants in the series of hexafluorocyclohexadienyl radicals derived by addition of the C-centred radicals to C6F6, Ingold [103] supposes that the cyclohexadienyl fragment undergo nonplanar deformation, the degree of which is probably determined by the steric requirements of R•.
The significant decrease of the 1H HFI constant in going from the non-fluorinated cyclohexadienyl radical or the pentafluorocyclohexadienyl radical with a –CH2– ring unit to the hexafluorocyclohexadienyl radical with a –CHF– ring unit reveals that the replacement of the geminal hydrogen atom by the fluorine atom, which hardly brings about the essential deformation of the cyclohexadienyl planar structure, is accompanied by the effect complying with this speculation[103]. If the above considerations are right, one should expect a lower ipso-F HFI value in going from the non-fluorinated cyclohexadienyl radical to the heptafluorocyclohexadienyl radical. Unfortunately, an attempt to photochemically add a fluorine atom to C6F6 in order to register the heptafluorocyclohexadienyl radical failed [103].
Authors [104] obtained ESR spectra of cyclohexadienyl radicals formed by C6H6 and C6F6 with aroyloxyl and aryl radicals generated by the direct UV-photolysis (high pressure mercury lamp, 1000W) of diaroylperoxides in cuvette of EPR-spectrometer at 25 °C. In the case of C6F6, the ESR spectra of cyclohexadienyl radicals formed both by aroyloxyl and aryl radicals were registered. For the cyclohexadienyl radicals derived by the addition of aroyloxyl radicals to C6F6, the ipso-F HFI constants are lower than in the analogous arylcyclohexadienyl radicals, obviously by the same reasons as considered above for hexafluorocyclohexadienyl radicals [103].
On heating (80°C) pentafluorobenzoyl peroxide in C6F6 or its photolysis (20 °C) an ESR spectrum of the perfluoro-1-benzoyloxycyclohexadienyl radical 18 was registered [101]. The structure was assigned on the ground the HFI parameters (αF1 = 82.0 G; αF2,6 = 22.4 G; αF3,5 = 6.5 G; αF4 = 36.8 G) which are close to those of the hexafluorocyclohexadienyl radicals with the geminal benzoyloxyl groups [103].
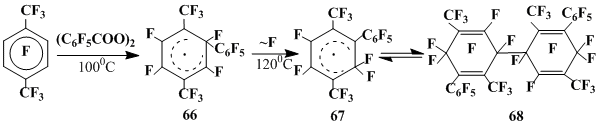
Since the rearrangement with fluorine migration in the course of the thermal decomposition of pentafluorobenzoyl peroxide in hexafluorobenzene occurs as a secondary process [19], we had used perfluoro-para-xylene as a substrate and a solvent, keeping in mind, that upon decomposition of pentafluorobenzoyl peroxide in octafluorotoluene [27] at 80 to 200°C only pentafluorophenyl radical participates in the reaction.
In so doing, at 90–100°C an ESR spectrum of 1-pentafluorophenyl-3,6-bis-trifluoromethyl-2,4,5-trifluorocyclohexadienyl radical 66 is registered. The αF1 value of 69.2 G for this radical is smaller than the corresponding αF(1) constants in both radical 18 (αF1 = 82.0 G) and phenylhexafluorocyclohexadienyl radical (αF1 = 107.3 G) [103]. This can be not only because of high electrone acceptance of the geminal pentafluorophenyl group in radical 66, but also due to its steric interaction with the 2-CF3 group. Thus, the possible out-of-plane deviation of the sp3 carbon and the distortion of the cyclohexadienyl fragment planarity result in a distinction of the αF3, αF4, and αF6 values of radical 66 from those typical for polyfluorinated cyclohexadienyl radicals [103, 104].
Appreciable changes in the ESR spectrum observed at the temperature elevation to 120–138 °C show the radical 66 transformation to a new radical. Two αF (1) constants with the close values of 50.4 and 50.0 G appear which indicates the isomerization of 66 via the germinal fluorine migration to the neighboring carbon atom to derive perfluoro-2,5-dimethyl-6-phenylcyclohexadienyl radical 67. The smaller values of αF(1) HFI constants of radical 67 compared to radicals with the –CFR– ring unit accords with the known dependence of these values on the steric interactions of the geminal substituent and its electronegativity [103]. The presence of two HFI constants testifies for a nonequivalence of the geminal fluorines that can be due to the above out-of-plane deviation of the sp3-carbon, but their closeness suggest this deviation insignificant and not concomitant with a nonplanarity of the cyclohexadienyl core. The g-factor value of 2.0037 of 67 is characteristic for radicals of this type [103], and the HFI constants αF(3) = 6.5 G and αF(4) = 38.2 G, unlike its precursor 66, are close to the values for hexafluorocyclohexadienyl radicals with a geminal non-fluorine substituent [103].
Radical 67 generation upon heating (100–130 °C) of dimer 68, which was isolated from the products of pentafluorobenzoyl peroxide decomposition in perfluoro-para-xylene at 140 °C, (Scheme 24) [101] additionally confirms its structure.
Scheme 24
Conclusion
Concluding the review, the most important peculiarities of polyfluoroarenes behaviour in the radical reactions should by outlined:
1. Preferable dimerization of the radical σ-complexes to form fluorinated tetrahydrobiaryls in the absence of defluorinating agents, whose role can be played both by special reagent and reactions products.
2. The lowered compared to non-fluorinated analogues reactivity of polyfluoroarenes with electrophilic radicals and their high reactivity with nucleophilic radicals.
3. Preferable meta-orientation in the reactions of C6F5X (X = CF3, C6F5) compounds with, electrophilic radicals (CF3•, C6F5•) and para-orientation of the nucleophilic radicals (CH3•, CH(CH3)OC2H5•) attack.
4. For OFN – preferable attack on the 1-position in the reactions with oxygen centered and alkyl radicals at low temperature, and the preferable formation of products of the attack on the 2-position with alkyl and aryl radicals at high temperature.
5. The previously unknown rearrangement via the fluorine 1,2-migration of polyfluorinated cyclohexadienyl radicals – the short living intermediates (radical σ-complexes).
6. A new type of the radical σ-complex stabilization such as their fragmentation to the corresponding aroxyl radicals as a specific peculiarity of the polyfluoroarene reactions with oxygen centered radicals.
References
Recommended for publication by Prof. V.E. Platonov
Fluorine Notes, 2012, 81, 1-2
