Received: June, 2011
Fluorine Notes, 2012, 80, 1-2
Synthesis and Application of Polyfluorinated Organic Derivatives of Boron and Silicon for Preparation of Iodonium, Bromonium and Xenonium Salts
V. V. Bardin
N.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry of the Siberian Branch of Russian
Academy of Sciences, Novosibirsk, Russia
E-mail: bardin@nioch.nsc.ru
Abstract: Preparation of polyfluoroorganylsilanes, boranes and borates and their use for synthesis of salts [RFXeFn][A], [RFR'FBr][A] and [RFR'FIFn][A] are considered.
Keywords: Organoborates, organoboranes, organosilanes, iodonium salts, bromonium salts, xenonium salts
List Of Contents
I. Introduction
II. Synthesis of Polyfluorinated Organylboranes, -borates and silanes
III. Synthesis of Salts of Polyfluorinated Derivatives of Bromine(III, V), Iodine(III, V)
and Xenon(II, IV)
I. Introduction
Belonging to "borderline" sciences, which include typical particularities of every main direction and specific properties typical for only present field of sciences is the particularity of chemistry of polyfluorinated organoelement compounds. That to a full extent can be referred to organic derivatives of polyvalent iodine and bromine. Preparative methods and reactivity of these compounds are studied quite well and presented in numerous reviews and fundamental monographs. Their polyfluorinated analogues have been studied considerably less, and just recently the information regarding them has been systematized in review [1]. The first reliable data on synthesis of organoxenon compounds appeared in 1989 when two groups of German scientists had independently published the preparation of [C6F5Xe][(C6F5)nBF4-n] salts [2, 3]. During research the availability of using the reactions of transfer of organic fragment R from silicon or boron atom onto xenon atom in XeF2 molecule, iodine or bromine in R'EF2 (E = Br, I; R' = F or organic group) had been revealed. In the present review we have analyzed the application of these reactions in preparative synthesis of perfluoroorganylxenon (II, IV) salts, symmetrical and asymmetrical bromonium (III) and iodonium (III, V) salts with perfluorinated organic groups bonded to halogen atom.
II. Synthesis of Polyfluorinated Organylboranes, –borates and –silanes
1. Boron Derivatives
Originally polyfluoroarylxenonium and –iodonium salts had been obtained from fluorocontaining triarylboranes. The synthetic procedure and particularly, the purification of these precursors was complex and the reaction products were salts of [ArnBF4-n]– anions. The usage of boranes RBF2 became an alternative route. A general scheme for preparation of RBF2 was elaborated and it included synthesis of polyfluororganyl(alkoxy)borates and polyfluoroorganyl(fluoro)borates. The key process stage is nucleophilic addition of polyfluoroorganyllithium (or polyfluoroorganylmagnesium bromide) to tris(alkoxy)borane:
RFM + B(OAlk)3 → M[RFB(OAlk)3]
M = Li, MgBr; RF = CnF2n+1, RCF=CF, CF2=CR, RC≡C, C6HnF5-n (n = 1-5)
Salts M[RFB(OAlk)3] can be isolated in their pure form [4-9], but to obtain derivatives of boric acid [8, 10] or fluoroborates [11-19] they are often used without isolation from solution.
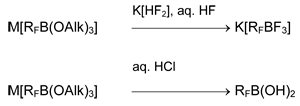
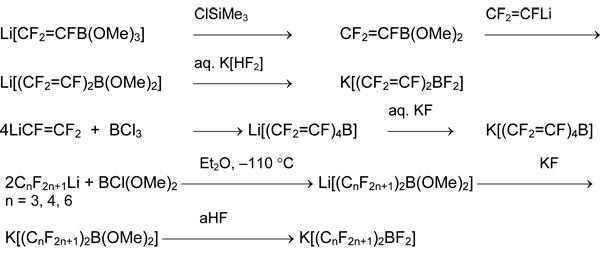
Based on this scheme, methods of preparation of organoboron compounds with 2-4 organic moieties at boron atom were developed [5, 18, 20].
The important property of organylfluoroborates is the ability to eliminate fluoride anion under the action of Lewis acid producing the corresponding organylfluoroboranes. Although the fluoride affinity of boranes RFBF2 in gaseous phase (pF-) is 8.51 (RF = C6F5), 8.0-9.0 (RF = CF3CF=CF), 8.90 (RF = CF3C≡C) and exceeds such one BF3 (7.88) [21], the bubbling of boron trifluoride into suspension K[RFBF3] in dichloromethane or polyfluorohydrocarbons results in formation of solution RFBF2 [11,12,16,17,22-24]. Presumably, the high energy of crystal lattice of precipitated K[BF4] is the driving force of the process.
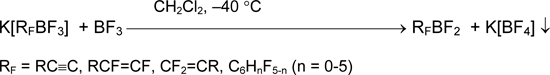
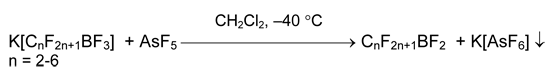
To defluoridate perfluoroalkyltrifluoroborates to perfluoroalkyldifluoroboranes (pF- ~ 9.6), the stronger Lewis acid like AsF5 is required [14].
Organyldifluoroboranes are well soluble in nonpolar solvents, and after decantation their solutions can be used for the further reactions. This method is applicable to boranes with hydrocarbon groups R and to their perfluorinated analogues [11, 12, 14, 16, 17, 22-24].
For more detailed discussion of reactivity of polyfluorinated fluoroboranes and fluoroborates see reviews [25, 26].
2. Silicon Derivatives
Polyfluoroarylsilanes ArFSiAlk3 and ArFSiF3 are the useful precursors. The reaction of polyfluoroaryl bromides and -iodides with ClSiAlk3 and tris(dialkylamino)phosphines is suitable for the synthesis of the former. Target products ArFSiAlk3 are formed fast at room temperature in aromatic hydrocarbons, ethers and esters (diethyl ether, diglyme, THF, ethyl acetate), dichloromethane or acetonitrile [27-31].

The use of trialkylgermanium, trialkyltin, and trialkyllead halogenides instead of ClSiAlk3 allows obtaining the corresponding organoelement compounds.

It is interesting, that in the presence of protic substrates the substitution of halogen atom by hydrogen (reduction) occurs [27, 32].

Hexafluorobenzene does not react with ClSiAlk3 and P(NAlk2)3. However, the substitution of one fluorine atom in hexafluorobenzene by the more electron-withdrawing substituent facilitates reaction of silyldefluorination and germyldefluorination like the substitution of bromine or iodine atoms [33, 34].
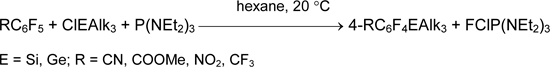
Such reactions do not occur in the line of the non-fluorinated or less-fluorinated aromatic compounds. But this approach is suitable for the synthesis of perfluorinated alkenylsilanes and cycloalkenylsilanes [35].
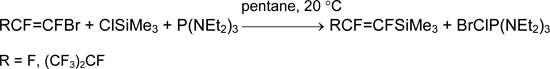
The attempts of pentafluorophenylation of SiCl4, GeCl4 and GeBr4 using P(NEt2)3 did not lead to the desired result. The alternative methods based on substitution of ethoxy groups in comparatively available silane C6F5Si(OEt)3 were elaborated for preparation of C6F5SiX3 (X = F, Cl) [36].
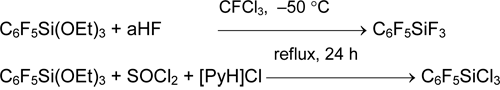
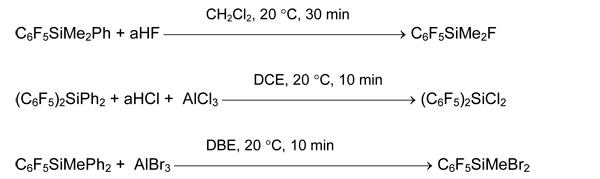
It was offered to obtain pentafluorophenyl-containing fluoro-, chloro- and bromosilanes of common formula (C6F5)nSiMemX4-n-m by cleavage of Si-Ph bond in the corresponding phenylsilanes with anhydrous hydrogen fluoride HF (aHF) or HSO3F (X = F), anhydrous HCl (aHCl) and AlCl3 (X = Cl) or AlBr3 in dibromomethane. The processes occur under mild conditions and with high yields [37, 38].
Aryl(fluoro)silanes and -germanes were prepared from the corresponding chloro- or bromoderivatives by the action of SbF3, NbF5 [36] or better, Na2[SiF6] [39].
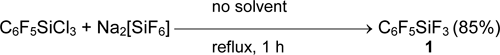
Silane 1 is weaker fluoride anion acceptor, than borane 2. Its fluoride anion affinity in gaseous phase (pF-) is 7.45 with respect to 8.51 of borane C6F5BF2. For comparison, the fluoride anion affinities of binary fluorides BF3 and SiF4 are 7.88 and 7.18, respectively. The substitution of fluorine atoms at heteroatom by alkyl groups significantly weakens the strength of Lewis acid. Thus, pF- value of silane C6F5Si(CH3)3 is 4.92 [21].
III. The Synthesis of Polyfluorinated Derivatives of Bromine (III, V), Iodine (III, V) and Xenon (II, IV)
1. Bromine (III) Derivatives
Pentafluorophenyldifluorobromane (4) was prepared by reaction of bromine trifluoride with C6F5SiMe3 (3) (1 equiv.) in dichloromethane in 58% yield. Bis(pentafluorophenyl)bromonium salt does not form even in excess of C6F5SiMe3 and in the presence of BF3•OEt2 [40].
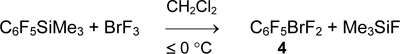
Pentafluorophenyltrifluorosilane (1) reacts with BrF3 in the same way at temperature below -60°C. The rate of the further reaction becomes significant at temperature above -30°C and as a result, salt [(C6F5)2Br][SiF5] is obtained in 92% yield.
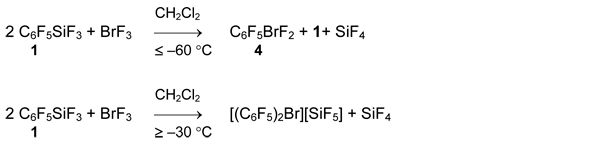
Polyfluoroorganylboranes are stronger fluoride anion acceptors (Chapter II.1) and their interaction with BrF3 in weakly coordinating solvents gave symmetrical bis(perfluoroorganyl)bromonium salts.
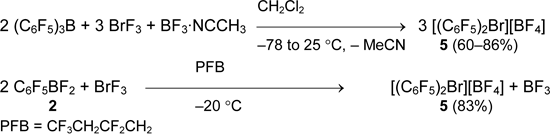
It should be noted, that salt 5 is formed even in equimolar reaction of pentafluorophenyldifluoroborane 2 with BrF3 while the formation of C6F5BrF2 was not registered at -60°C [40].
The method of preparation of bis(pentafluorophenyl)bromonium tetrafluoroborate was successfully applied for the synthesis of representative series of bis(perfluoroorganyl)bromonium salts containing alkenyl and alkynyl groups.

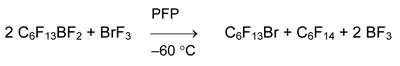
The attempt to obtain or detect bis(perfluorohexyl)bromonium salt in the reaction of C6F13BF2 with BrF3 failed.
The reactions of aryldifluorobromanes with aryltrifluorosilanes were used for preparation of asymmetrical fluorine-containing bis(aryl)bromonium salts [41].
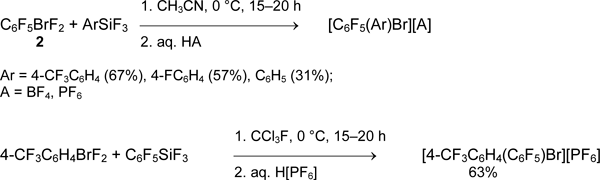
It can be expected, that aryldifluorobromanes will also react with alkenyl- and alkynyltrifluorosilanes, though the latter are low-available and there is no information on such research.
On the contrary, polyfluorinated alkenyldifluoroboranes and alkynyldifluoroboranes are easily available (Chapter II.1). Their reactions with bromanes ArBrF2 gave the corresponding perfluoro(aryl)(alkenyl)bromonium and perfluoro(aryl)(alkynyl)bromonium salts.
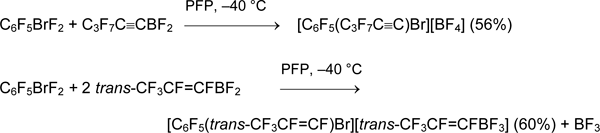
Attempts to synthesize perfluorinated phenyl(hexyl)bromonium and (pentafluorophenyl)(1H,1H,2H,2H-tridecafluorooctyl)bromonium salts failed.
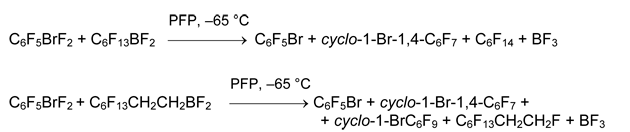
It should be noted, that organyl(fluoro)bromonium salts [RBrF][A] are not registered in the above reactions and are not described in literature. The interaction of C6F5BrF2 with BF3 in weakly coordinating solvent (SO2FCl) and basic solvent (CD3CN + CD2Cl2) was studied [42]. In the former case the fast decomposition of 4 with formation of C6F5Br and products of latter was observed. In second case a complex [C6F5BrF •(NCCH3)n][BF4] (19F NMR) was found, which decomposed at heating up the solution to 22°C.
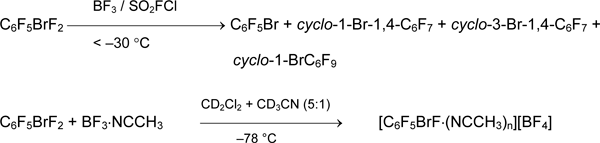
2. Bromine (V) Derivatives
In contrast to bromine (III) derivatives, organylbromane (V) salts are not described, but covalent arylbromanes ArBrF4 are known. Pentafluorophenyltetrafluorobromane was obtained in 66% yield by reaction of pentafluorophenyltrimethylsilane with bromine pentafluoride in the presence of MeCN. Bis(pentafluorophenyl)dimethylsilane and 4-trifluoromethylphenyltrimethylsilane react in the same way [43].
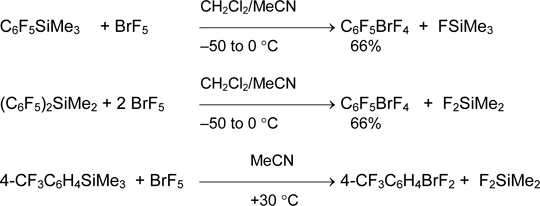
The replacement of aryltrimethylsilane by aryltrifluorosilane (stronger Lewis acid) does not alter the reaction direction and the character of product. The only products of aromatic ring fluorination are formed in excess of base as well as without it [43, 44].
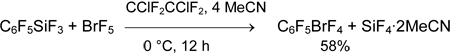
Bromane 3-CF3C6H4BrF4 was synthesized from BrF5 and complexes 3-CF3C6H4SiF3•2Py or (3-CF3C6H4)2SiF2•2Py using this method. At the same time, the corresponding aryltetrafluorobromanes FC6H4BrF4 were formed from 2-FC6H4SiF3•2Py and 4-FC6H4SiF3•2Py in very low yield [43].
![]()
3. Iodine (III) Derivatives
The convenient preparative route to aryl(polyfluoroalkyl)iodonium and alkynyl(polyfluoroalkyl)iodonium salts was offered by Zhdankin [45-47]. It includes the treatment of polyfluoroalkyliodine derivatives (6) with aryltrimethylsilane or alkynyltrimethylsilane in dichloromethane.
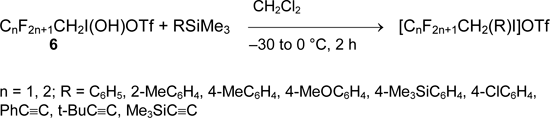
Perfluoroalkyliodine (III) derivatives (7) are the less reactive and form the corresponding aryl(perfluoroalkyl)iodonium salts only in the presence of Me3SiOTf. Alkynyltrimethylsilanes do not react with them.
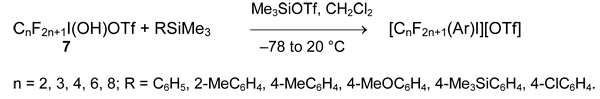
This method is limited by using of silanes RSiMe3 with electron-donating substituent R. The more general method is based on interaction of organyldifluoroiodanes RIF2 with triarylboranes or better organyldifluoroboranes R'BF2. In the first case aryl(polyfluoroaryl)iodonium salts are formed [48-50].
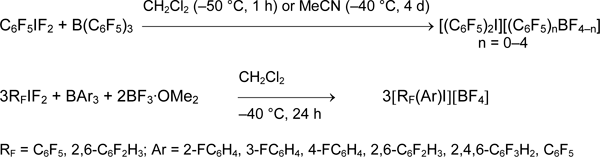
Using boranes, symmetrical and asymmetrical salts with aryl, alkenyl, and alkynyl groups in different combinations were formed. Moreover, different pairs of reagents RIF2/R'BF2 or R'IF2/RBF2 can be used in depend on their availability [4,21,51, 52].
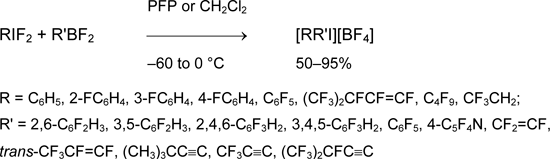
Using this method, perfluorinated aryl(alkyl)iodonium salt was obtained for the first time [53].

4. Iodine (V) Derivatives
Silane 1 does not react with excess of neat IF5 and its solutions in CH2Cl2, fluorochlorocarbons or in anhydrous HF [36, 54], but in acetonitrile it produces pentafluorophenyltetrafluoroiodane (8) in quantitative yield [54]. This iodane was also obtained from tetramethylammonium pentafluorophenyltetrafluorosilicate [55].
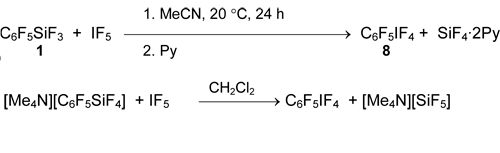
Pentafluorophenyltrifluoroiodonium salt 9 does not form here, but it can be generated by the action of SbF5 in SO2FCl [42, 56].
![]()
Perfluoroalkyltrifluoroiodonium salts are not described and apparently they are very unstable. For example, the treatment of perfluorohexyltetrafluoroiodane with arsenic pentafluoride leads to carbon-iodine bond cleavage and formation of perfluorohexane with quantitative yield [42].

It is interesting, that the action of [IF4][SbF6] on silane 1 in fluorosulfonic acid results in formation of bis(pentafluorophenyl)iodonium salt instead of salt 9 [36].
![]()
Borane 2 reacts with iodine pentafluoride in CH2Cl2 at 20°C very slowly producing a mixture of C6F5IF4, [(C6F5)2IF2][BF4] and by-products (C6F5I, C6F5H, CH2ClF). Its reaction with iodane 8 occurs faster and leads to bis(pentafluorophenyl)difluoroiodonium tetrafluoroborate [57].

5. Approaches to Preparation of Iodine (VII) Derivatives
Inorganic iodine (VII) compounds are the less studied relative iodine (III) or iodine (V) derivatives, and organic iodine (VII) compounds are not described at all. Recently some approaches to RFIF6 based on using of organoboron and organosilicon synthones and IF7 are studied [58]. In contrast to above considered cases, the reaction of iodine heptafluoride with C6F5SiMe3 or C6F5SiF3 in PFP produces only products of fluorine addition to aromatic ring. The substitution of silicon atom by iodine takes place in the reaction of [Me4N][C6F5SiF6] with IF7 but the reaction products are iodine (V) derivatives.
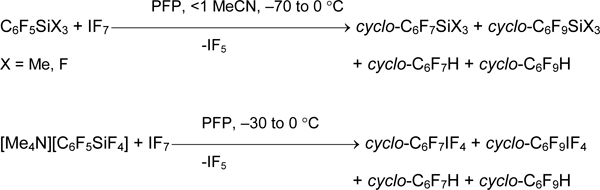
The reaction of IF7 with borane 2 and even with perfluorodiborylbenzene, 1,4-C6F4(BF2)2, which contain two strong electron-withdrawing groups in tetrafluorophenyl ring [σI (BF2) = 0.15, σR (BF2) = 0.30, σm (BF2) = 0.45, σp (BF2) = 0.37] [11] proceeds in similar way.
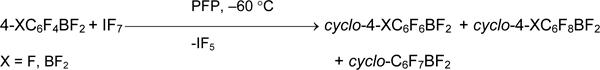
The analysis of relative reaction rates of C6F5SiX3 and C6F5BF2 with HalFn (Hal = Br, I; n = 3, 5) proved their decrease when n increases. It is one of more possible causes for the absence of bromine or silicon atom substitution for I(VII) atom. Notably the rate of iododeborylation by action of IF7 is lower, than the rate of parallel polyfluoroaromatic ring fluorination reaction.
6. Xenon (II, IV) Derivatives
First publications on successive preparative synthesis of compounds with carbon-xenon bond were published in 1989 [2, 3]. Shortly before that publications communication [59] on reaction of perfluoroaryltrimethylsilanes ArFSiMe3 with xenon difluoride over cesium fluoride at 20°C (heterogeneous reaction) appeared. Besides xenon and fluorotrimethylsilane, products were arenes and biaryls. The reaction of XeF2 with Cs[C6F5SiF4] goes analogously [55].
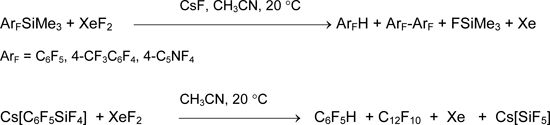
The formation of organoxenon compounds was not proved. The causes became clear later when properties of unambiguously prepared and characterized salts (like [C6F5Xe][AsF6]) and covalent compounds (like C6F5XeF or (C6F5)2Xe) had been studied. Indeed, organic compounds of two-coordinated xenon are significantly less stable, than the respective salts, and they dissociate in acetonitrile on radical mechanism producing C6F5H and C12F10 [60, 61]. When the reaction of C6F5SiMe3 and XeF2 in the presence of [Me4N]F was performed by heating homogeneous solution in MeCN-EtCN from -60 to -40 °C with further fast cooling, then C6F5XeF and (C6F5)2Xe was registered with 19F and 129Xe NMR spectroscopy. But xenon compounds with trifluoromethyl groups are unstable even at low temperature. Thus, under these conditions only hexafluoroethane, xenon and FSiMe3 were formed from CF3SiMe3 [62].
Similar approach was used for preparation of bis(pentafluorophenyl)xenon from pentafluorophenylfluoroxenon 10 and silanes 1 or 3. Unexpectedly, the formation of this compound was found in the reaction of 10 with silanes CF3SiMe3 and CF2=CFSiMe3 in the presence of [Me4N]F [63].
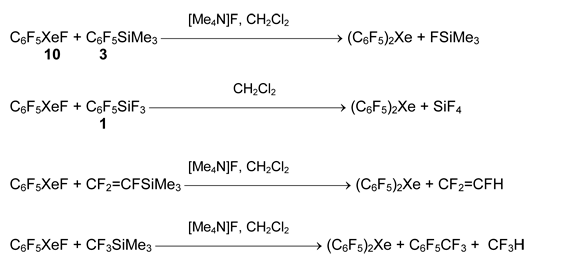
Practically useful results in preparative synthesis of organoxenon compounds were obtained when boron compounds were used as organic group carriers. Originally triarylboranes were used, but because of relatively high nucleophilicity of [(XC6H4)nBF4-n] anions, these salts decompose in solution at -60 to -20 °C.
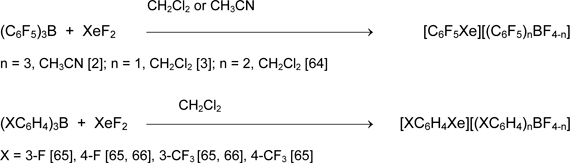
The better result was obtained in the presence of boron trifluoride etherate. The role of the latter consists in replacement of relatively good fluoride anion donors, [ArnBF4-n]-, by the low-nucleophilic anions [BF4]-, which significantly increases the stability of [ArXe][A] salts.
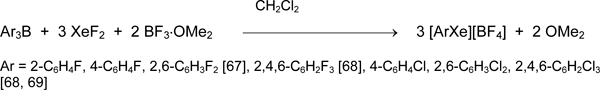
A significant progress in research of organoxenon compounds was achieved after usage of organyldifluoroboranes RFBF2 as sources of RF. A simple and convenient preparative synthetic method to polyfluororganylxenon (II) tetrafluoroborates containing aryl, alkenyl, and alkynyl groups at xenon atom was elaborated [12, 16, 17, 70-75].
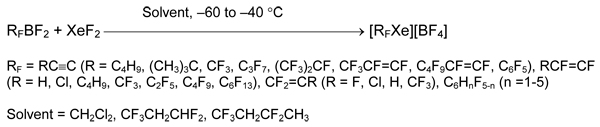
This method has some limitations. Thus, reaction of XeF2 and 4-tetrafluoropyridyldifluoroborane gave presumably complex 4-C5NF4BF2•XeF2 [21] instead of expected tetrafluoropyridylxenonium (II) salt [21]. Alkylxenonium derivatives remain unknown: reaction of C6F13BF2 and XeF2 leads to perfluorohexane, xenon, and boron trifluoride, whereas from C6H13BF2 and XeF2 mainly hexyl fluoride was formed.
The reaction of borane 2 with xenon tetrafluoride proceeds significantly slower than with XeF2 and produces pentafluorophenyldifluoroxenonium salt [76].

The alternative approach consists in electrophilic substitution of heteroatom of very good leaving group. In regards to that, a reaction of K[C6F5BF3] and XeF2 in anhydrous HF is representative, because the fluorine addition to aromatic ring proceeds simultaneously with the substitution of boron atom by xenon [77].
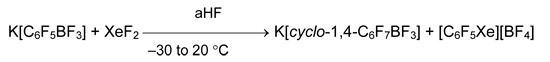
Detailed analysis of contemporary state of chemistry of organoxenon compounds see reviews [60, 61].
References
Recommended for publication by Prof. V.E. Platonov
Fluorine Notes, 2012, 80, 1-2
