Fluorine Notes, 2011, 79, 1-2
ELECTROPHILIC REACTIONS, CARBOCATIONS AND CATIONOID SKELETAL TRANSFORMATIONS IN THE SERIES OF POLYFLUOROARENES
V.M.Karpov, T.V.Mezhenkova, V.E.Platonov
N.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry of the Siberian Branch of Russian Academy
of Sciences, Novosibirsk, Russia
E-mail: karpov@nioch.nsc.ru
Abstract: In this work the data regarding generation and properties of polyfluorinated arenium, benzyl, indenyl and fluorenyl cations has been generalized. The mechanisms of cationoid skeletal transformations of polyfluorinated benzocycloalkenes and their carbonyl derivatives passing with contraction, expansion and opening of alicyclic fragment of substrate have been discussed as well.
Keywords: polyfluoroarenes, benzocycloalkenes, electrophilic reactions, carbocations, cationoid skeletal transformations.
Carbon framework of polyfluoroaromatic compounds depleted by electrons due to strong inductive effect of fluorine atoms is deactivated towards the attack of electrophylic reagents. At the same time due to the lone-pair electrons fluorine atoms can participate in stabilization of formed carbocations formed according to mechanism of p,π-coupling.
The possibility of electrophilic substitution of hydrogen atom for incompletely fluorinated aromatic compounds had been shown for the first time using the example of nitration of 1, 2, 3, 5-tetrafluorobenzene [1], as well as of iodation, bromination and sulfonation of pentafluorobenzene [2, 3]. The transformations of perfluoroarenes which are caused by the unknown for aromatic compounds ipso-attack of electrophyl on the position occupied by fluorine atom had been discovered at the example of nitrating reagents attachment to octafluoronaphtalene [4,5] (Scheme 1).
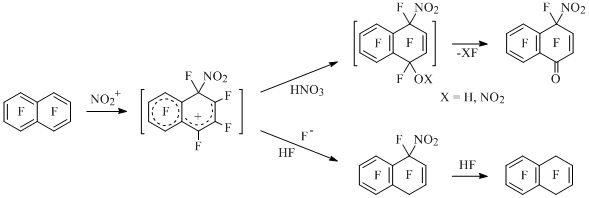
Scheme 1
The reactions of different polyfluoroarenes with nitrating agents had been thoroughly studied and along with the other data on interaction of polyfluoroaromatic compounds with electrophiles were united in the review [6] and monography [7].
At that it was impossible to fix in reaction and generate using another method the arenium ions responding to the attachment of cation nitronium to the polyfluoroarene and containing at the same time fluorine atom and nitro-group at sp3-hybridized carbon [7,8]. At the same time similar cations containing the geminal CFC1-unit were generated, including ones generated during electrophylic chlorination of polyfluorobenzenes and polyfluoronaphtalenes at low temperature in Cl2-SbF5-SO2ClF system [9-11] (Scheme 2), for example:
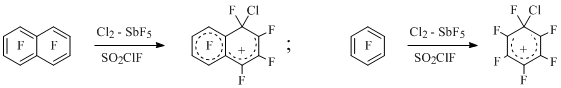
Scheme 2
Besides that, during chlorination of 1-H-heptafluoronaphtalene a rare case of naphthalenium ion formation has been registered, that ion is an intermediate at substitution of hydrogen atom and it produces 1-chloroheptafluoronaphtalene [11] at methanol treatment (Scheme 3).
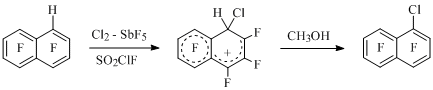
Scheme 3
The first perfluorinated arenium ions have been generated by abstraction of the fluorine anion from the perfluorodehydroaromatic compounds using antimony pentafluoride [12] (Scheme 4).
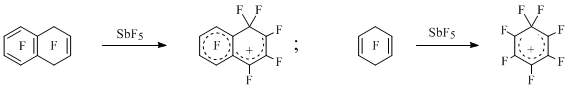
Scheme 4
A big number of stable polyfluorinated benzenium, naphthalenium and antracenium ions containing substituents in different positions had been generated using that method along with electrophilic halogenation and methylation (CH3F-SbF5) of polyfluoroarenes as well as protonation (HSO3F-SbF5) of polyfluorocyclohexadienons. Summarized data on structure of these cations, their electronic structure, relative stability and chemical properties had been analyzed in the review [8] and monography [7]. At that, we should point out an unexpected fact of forming of methylheptafluoronaphtalenes (yield 40-50%) during methylation of octafluoronaphtalene [13] (Scheme 5).
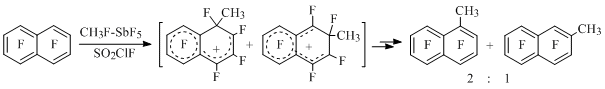
Scheme 5
As a result of elimination of the fluorine, chlorine or bromine anion from position 1 of polyfluoroindenes under the influence of excess of antimony pentafluoride in SO2 or SO2ClF a set of polyfluorinated indenyl cations [14-19] (Scheme 6) was generated , for example:
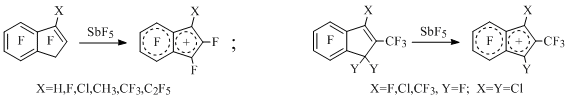
Scheme 6
The π-system of polyfluoroindenyl cations carbon framework as well as that of antiaromatic indenyl cation [20-23] contains 4n (8) electrons. According to that they must be referred to as anti-aromatic. However, as opposed to indenyl cation in its fluorine containing analogues the interaction of fluorine atoms p-electrons with the π-system of carbon framework along with the π-σ-interactions exaggerate the case. Along with that a criterion had been taken in to estimate their degree of (anti)aromaticity, which was used in the works by Dewar [20] and Breslow [21-23] and which main idea is the comparison of energy characteristics of cyclic and corresponding open-chain ions which are not isomers. At that we should take into account different number of σ-bonds, as well as their compression index in cyclic and open-chain ions [20] (Scheme 7).

Scheme 7
In the work [16] within the range of present criterion it had been offered to use charged and noncharged compounds able to formally produce isomers with both cyclic and "open" conjugated chain (Scheme 8), for example:
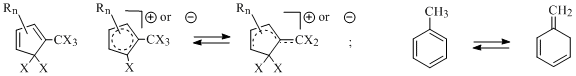
Scheme 8
Using such models allows direct experimental studying of relative stability of cyclic and "open" systems. From the point of quantum-chemical calculations view the advantage of these models is the possibility for direct comparison of total isomers’ energies not placing the mistake at account of attaching of additional atoms for compounds with open conjugated chain [24].
It happened to be so, that compare to antiaromatic indenyl cation which relative stability is lower than the one of its analogue with an open π-system the generated in solution perfluoroalkylindenyl cations and polyfluoroalkylindenyl cations with chlorine atoms in five-membered cycle proved to be more stable in terms of thermodynamics than isomeric to them alkylidenindanyl ions with open π-system [16, 19, 25].
Thus, cation 1which exists in temperature range from –60 to +140°C is generated from perfluoro-2-methylindene in the system of SbF5-SO2 or SbF5. At keeping the salt solution of this cation in antimony pentafluoride at room temperature for more than two years not a single change is happening in NMR 19F spectrum of cation.
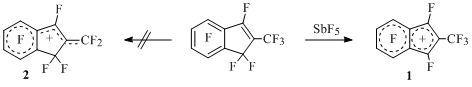
Scheme 9
Perfluoro-1-alkylindene cations 3 and 4 and their "open" analogues 5 and 6 (E, Z-isomers) are generated and it is proved, that the first mentioned are more stable than the last ones in terms of thermodynamics in spite of relative stability of neutral predecessors [19] (Scheme 10).
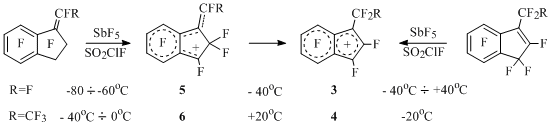
Scheme 10
Greater thermodynamic stability of cyclic cations compare to their "open" isomers is also observed in the case of chloro- and dichloroperfluoromethylindenyl cations. Thus, corresponding methyleneindanyl cations, which are isomerized into 3-chloroperfluoro-1-methylindenyl cation (7) [25] at 0°C, are generated from E,Z-isomers of 1-(fluorochloromethylene)octafluoroindan under the influence of antimony pentafluoride at low temperature [25] (Scheme 11).
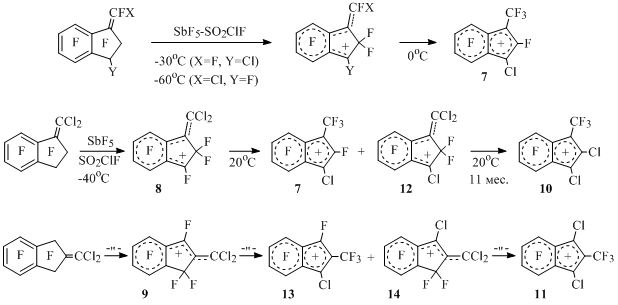
Scheme 11
Analogously cations 8 and 9 are generated from 1- and 2-dichloromethyleneoctafluoroindanes, at room temperature these cations gradually transform into dichloroperfluoromethylindenyl cations 10 and 11 through the intermediate formation of indenyl (7 and 13) and methyleneindanyl (12 and 14) cations [25] (Scheme 11).
Polyfluoroindenyl cations greater thermodynamic stability compare to their isomers with open π-system gave a reason to doubt the antiaromatic nature of such cations and assume their aromatic character [16,19]. At that they did not exclude, that in some other cases the introducing of substituents with strong +M or –M effect into anti-aromatic compound molecule would allow "to convert" that compound into aromatic one or at least low its degree of antiaromaticity.
The results of quantum-chemical study of fluorine containing cyclopentadienyl, indenyl and β-naphtindenyl cations do not come into contradiction with that [24]. The degree of their (anti)aromaticity had been estimated using MNDO method within the range of Dewar-Breslow criterion using difference of enthalpies of formation of isomeric cations with cyclic and open π-system: Δ=ΔHf(cycl.)-ΔHf(open)as aromaticity index. It had been proved, that the degree of aromaticity depends to a great extent on both carbon framework and presence and location of fluorine atoms inside it. At that for methylcyclopentadienyl cations the Δ value is always positive (antiaromatic cations), and for methylindenyl and methyl-β-naphtindenyl cations the alteration of sign is possible under the influence of fluorine atoms, e.g. the cyclic π-system can become energy-wise more efficient than the open one [24] (Scheme 12), for example:
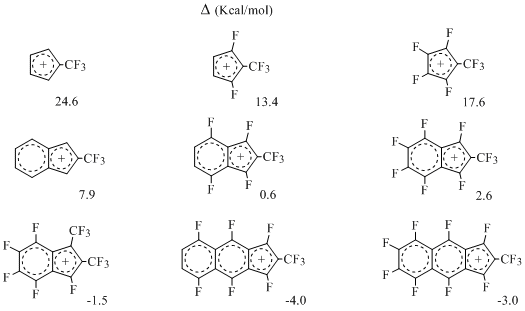
Scheme 12
The linear correlation dependence between the Dewar-Breslow aromaticty index (Δ) and the energy of lowest singlet-singlet excitation for cations with cyclic π-system had been found. Including analogues points for aromatic toluene and methylcyclopentadienyl anion into statistical manipulation led to linear dependence with close parameters but with higher correlation coefficient. Existence of such dependence allows considering the magnitude of energy of lowest singlet-singlet excitation in compounds with cyclic π-system as universal characteristic of their aromaticity degree incl. cyclic systems which do not possess open conjugated chain isomers [24].
Stable perfluorinated 9-methylfluorenyl, fluorenyl and 9-phenylfluorenyl cations containing 4n (12) electrons in π-system had been generated besides polyfluoroindenyl cations. Salt solution of the first cation had been obtained by elimination of the fluorine anion from corresponding fluorene under the action of antimony pentafluoride [26] (Scheme 13).
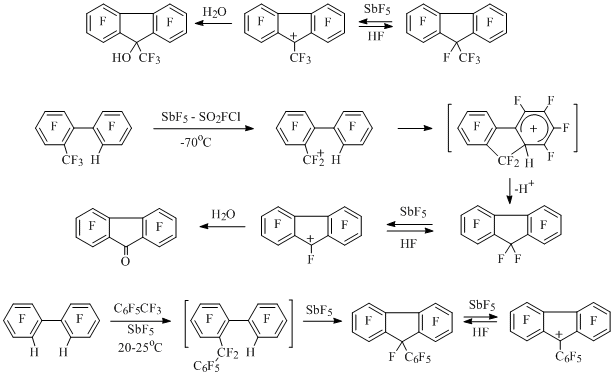
Scheme 13
In two last cases the fluorene frame was formed as a result of electrophylic alkylation of biphenyl derivatives. Thus, at dissolving of 2-trifluoromethyl-2'-Н-octafluorobiphenyl in SbF5-SO2ClF biphenylmethyl cation is being generated at low temperature, it undergoes intramolecular cyclization forming perfluorofluorene and later perfluorofluorenyl cation [27]. During interaction of 2,2'-H,H-octafuorobiphenyl and heptafluorobenzyl cation generated from octafluorotoluene in antominy pentafluoride a two-step alkylation of biphenyl takes place forming salt solution of perfluoro-9-phenylfluorenyl cation, which after anhydrous hydrogen fluoride treatment produces perfluoro-9-phenylfluorene [27] (Scheme 13).
Earlier pentafluorophenylmethyl cation had been generated by abstraction of the fluorine anion from pentafluorophenylfluoromethane under the influence of antimony pentafluoride [28] (Scheme 14). Corresponding benzyl cations had been generated from pentafluorophenyldifluoromethane, octafluorotoluene and 4-X-heptafluorotoluenes (X=H,F,Cl,Br,CH3) under the influence of SbF5 and using the same method [29]. These cations interact with tetrafluorobenzenes and pentafluorobenzene in the medium of antimony pentafluoride forming diphenylmethyl cations, which do not react with the excess of polyfluorobenzene [30] (Scheme 14).
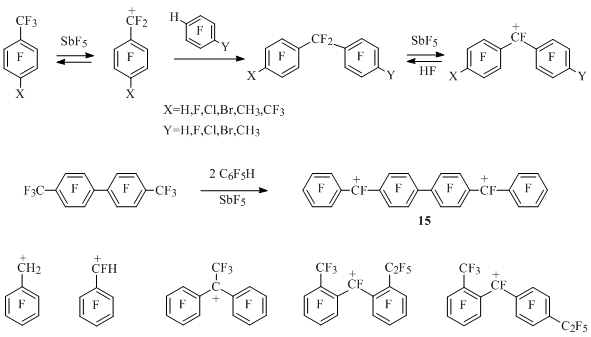
Scheme 14
The solution of perfluoro-p-xylene in SbF5 doesn't contain perfluoro-p-methylbenzyl cation in the concentration sufficient for observation using NMR 19F method [29], though in small concentration it apparently appears, which the formation of diarylmethyl cations (X=CF3, Y=F, CH3) in the reaction of perfluoro-p-xylene with pentafluorobenzene and 2,3,5,6-tetrafluorotoluene in the medium of SbF5 can indicate [30] (Scheme 14).
Perfluoro-4,4’-dimethylbiphenyl in the medium of antimony pentafluoride also doesn’t produce mono- or dications in noticeable concentration, though it interacts with two moles of pentafluorobenzene forming dication 15 [30] (Scheme 14).
By elimination of the fluorine anion from corresponding perfluorodiarylmethanes under the action of antimony pentafluoride diarylmethyl cations containing perfluoroalkyl groups at cation centre [30] or in resonance positions of both rings [31] had been generated (scheme 14).
Polyfluorobenzocycloalken-1-yl cations can be as well referred to benzyl type cations. Thus, 1-chloroctafluorindan-1-yl cation [32] is formed from 1,1-dichloroctafluoroindan in antimony pentafluoride or at heating of perfluoroindan with the mixture of SbCl5-SbF5, which have been taken in the molar ratio ~1:0.25:3 (Scheme 15). Perfluorobenzocyclobuten-1-yl cation [33,34] is generated at dissolving of perfluorobenzocyclobutene in antimony pentafluoride. Perfluorinated indan and tetralin under analogous conditions don’t produce similar ions in noticeable concentration [34], though they as well as benzocyclobutene react with pentafluorobenzene forming perfluoro-1-phenylbenzocycloalken-1-yl cations [35] (Scheme 15).
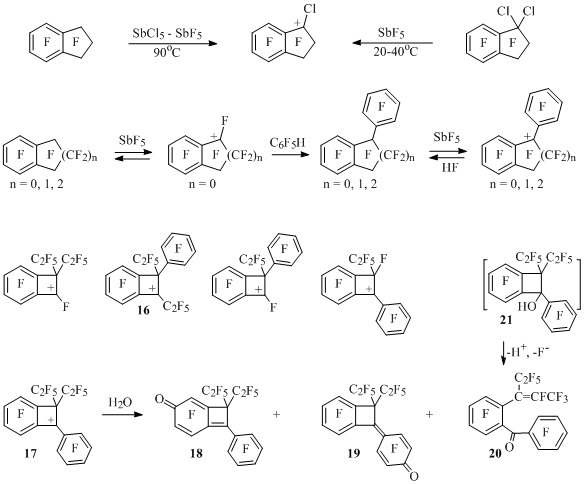
Scheme 15
Besides that, polyfluorobenzocyclobuten-1-yl cations [36] showed at Scheme 15 have been generated likewise. At interaction of cation 17 with water compounds 18-20 are formed, they respond to the attachment of water not only to the charge centre (alcohol 21) but also to the other carbon atoms located in resonance positions of ion 17. Benzophenone 20must be formed from alcohol 21, in whichthe opening of four-membered cycle occurs according to haloform reaction type. The same picture can be observed in case of cation 16 [36]. Perfluorinated 1-phenyl-2-methylbenzocyclobutene [37], 1-phenyl-3-ethylindan, 1,3-diphenyl-3-ethylindan and 1-phenyl-4-ethyltetralin [38] formed in the reaction of perfluoroalkylbenzocycloalkenes with pentafluorobenzene in the presence of SbF5, in the solution of antimony pentafluoride must also exist in the form of 1-phenylbenzocycloalken-1-yl cations, as during hydrolysis of solutions corresponding alcohols and ketons are being formed (Scheme 16).
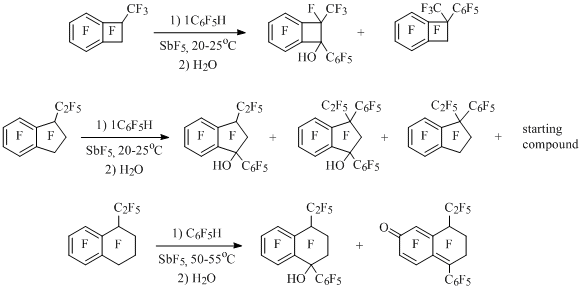
Scheme 16
Interaction with water leading to acids, carbonyl or hydroxy derivatives is typical for all long-lived cations considered above (benzyl, indenyl, benzenium etc.) At that, alongside with hydrolysis the attachment of fluorine anion to cation can occur forming corresponding perfluorinated compound. Thus, for example, at hydrolysis of salt solution of heptafluorobenzyl cation prepared from octafluorotoluene and SbF5, taken in molar ratio of 1:5, pentafluorobenzoic acid is obtained with the yield of 48%. At that, 19% of octafluorotoluene is being returned [39] (Scheme 17).
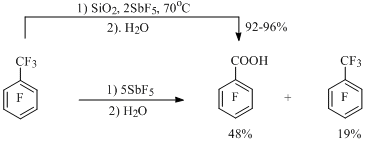
Scheme 17
During carrying out of octafluorotoluene and SiO2/SbF5 reaction pentafluorobenzoic acid forms with the high yield [40], at that using smaller amount of antimony pentafluoride than in the previous case (Scheme 17). This method allows synthesizing both mono- and di-carbonyl derivatives of perfluorinated alkylbenzenes and benzocycloalkenes, including the ones, which do not produce cations in noticeable concentrations [40-42] (Scheme 18), for example:
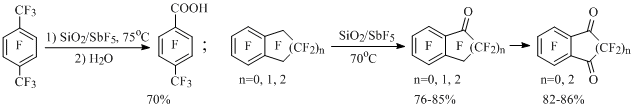
Scheme 18
In the reaction of perfluoro-1-ethylindan (R1 = F, R2 = C2F5) with SiO2/SbF5 the corresponding indanone is formed with small yield in the mixture with other compounds [43]. The substitution of SiO2 by trifluoroacetic acid as a source of nucleophilic oxygen allows carrying out the reaction under more mild conditions and obtain carbonyl derivatives of perfluorinated alkylbenzenes and benzocycloalkenes with high yield [44], for example:
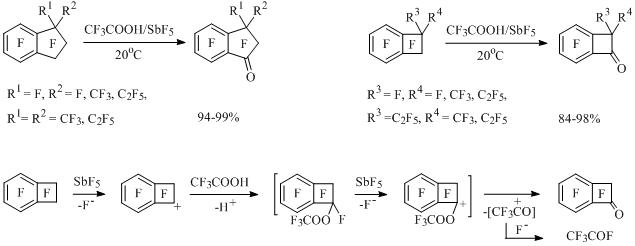
Scheme 19
The reactivity of perfluorobenzocycloalkenes at interaction with SiO2/SbF5 and CF3COOH/SbF5 coincides with relative stability of benzocycloalken-1-yl cations and lowers at trasition from benzocyclobutene to indan and tetralin.
Polyfluorobenzocycloalken-1-yl cations can act as alkylating agents towards fluorolefines. Thus, perfluoro-1-phenylbenzocyclobuten-1-yl cation reacts with tetrafluoroethylene forming perfluoro-1-phenyl-1-ethylbenzocyclobutene [36]. In the reaction of perfluorinated benzocyclobutene, indan and tetralin with tetrafluoroethylene in the presence of SbF5 perfluoro-1-ethylbenzocycloalkenes are obtained, which later are transformed into diethylderivatives.The reactivity of benzocycloalkene decreases by the size enlargement of its alicyclic fragment. At that, perfluoro-1,4-diethyltetralin is formed from tetralin, the mixture of perfluoro-1,1- and -1,2- diethylbenzocyclobutenes is formed from benzocycbutene at approximately even ratio, and from indan – only perfluoro-1,1-diethylindan [34, 45] (Scheme 20).
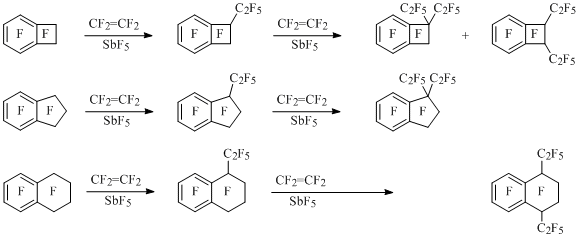
Scheme 20
Due to destabilizing influence of perfluoroalkyl group located by the charge centre in benzocycloalkenyl cations B (Scheme 21), we should be expecting formation of 1,2-,1,3- and 1,4-diethylderivatives from benzocyclobutene, indan and tetralin with cations A participating. The analysis of electronic and steric factors influencing the reactivity and relative stability of A and B carbocations made it possible to explain the obtained results [34].
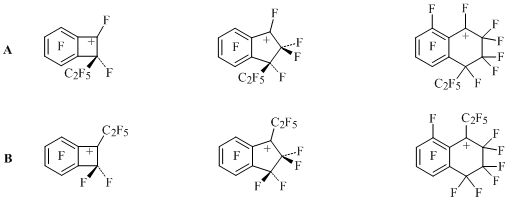
Scheme 21
Similar regularities are observed in catalyzed by SbF5 reaction of benzocyclobutene with trifluorochloroethylene and perfluoropropylene, of perfluoro-1-methylbenzocyclobutene with tetrafluoroethylene and perfluoropropylene [33], and also perfluorinated indan, benzocyclobutene and 1-methylbenzocyclobutene with 1,2-difluorodichloroethylene [46]. Perfluorovinylbenzocycloalkenes [46] are synthesized by dechlorination of polyfluorobenzocycloalkenes and difluorodichloroethylene interaction products.
Cationoid skeletal transformations of perfluorinated benzocycloalkenes and their carbonyl derivatives unknown for perfluorocarbons have been discovered and currently being studied in N.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry. Thus, at heating of 2-bromoheptafluoronaphtalene with antimony pentafluoride up to 300°C with simultaneous distillation of formed products a complex mixture is being formed containing 20-25% of perfluoro-1-methylindan (22). The authors suppose, that in the beginning the fluorination of naphthalene occurs forming 2-bromoperfluorotetralin (23), from which perfluorotetralin-2-yl cation is generated (24), and in it six-membered cycle is being contracted to five-membered one [47] (Scheme 22). In the work [48] it is shown that under analogous conditions the mixture containing methylindan was formed from 2-bromotetralin 23 as well as from 1,2- and 2,3-dibromodecafluorotetralins.
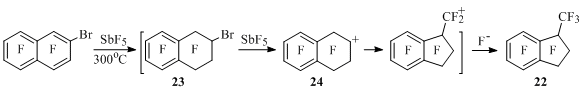
Scheme 22
During the transition from bromo- to chloropolyfluorotetralins the yields of contraction products of six-membered ring to five-membered one are noticeable increasing. At that in the reaction of 2-chloropolyfluorotetralins 25 containing no more than one chlorine atom at one sp3-hybridized carbon atom indan 22 and chloroindan 26 are obtained. Along with that partial or total substitution of chlorine atoms by fluorine atoms occurs in the initial compound 25. At accumulation of chlorine atoms in tetralin 25 the indan 22 yield decreases from 63% to 22%, and the yield of chloroindan 26 increases from 8% to 19% (Scheme 23).
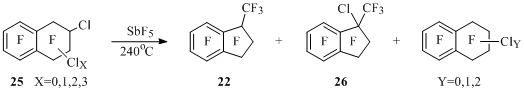
Scheme 23
Under analogous conditions 1,1,4,4-tetrachlorooctafluorotetralin and perfluorotetralin do not produce ring contraction products [48], though during prolonged heating of perfluoroteralin with SbF5 (200°C, 34 h) indan 22 is being formed along with perfluoro-2-isopropyltoluene [49].
Using the example of 2-chloroperfluorotetralin 27 and antimony pentafluoride interaction they have offered a contraction mechanism of six-membered cycle to five-membered one containing generation of tetralin-2-yl cations 24, 28, in which the cation centre attacked aromatic ring and arenium ions 29 formed later were isomerized into indanylmethyl cations 30. They attached fluorine anion producing indanes 22 and 26 (Scheme 24).
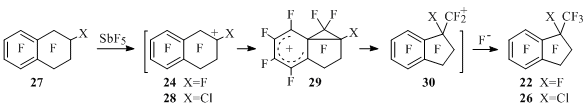
Scheme 24
By a separate experiment it has been established, that under the influence of antimony pentafluoride product 26is partially being transformed into indan 22 [48]. However, in the work [50] it was shown, that the indan 22 itself could produce chloroindan 26 under the reaction conditions. In connection with that the formation of chloroindan 26 can’t serve as a reason for approval of discussed in works [47,48] (Schemes 22,24) mechanism of contraction of six-membered ring to five-membered one, according to which carbon atom located in α-position of tetralin appears in trifluoromethyl group of the indan being formed (Scheme 24).
Perfluoro-1-ethyltetralin (31) at heating together with SbF5 isomerizes into ethylteralin 32, in which six-membered ring contracts to five-membered one forming methylethylindan 33. At higher temperature it transforms into trimethylindan 34. In a similar way perfluoro-1,4-diethyltetralin (35) isomerizes into diethyltetralin 36 under the influence of SbF5 and then six-membered alicyclic fragment contracts to five-membered or even four-membered one forming isomeric trialkylindan 37 and tetraalkylbenzocyclobutene 38 [50,51] (Scheme 25).
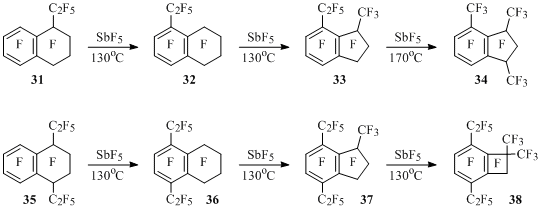
Scheme 25
The contraction of alicyclic fragment in tetralins 32 and 36 containing electron-withdrawing C2F5 groups proceeds surprisingly easier (130°C) compared to perfluorotetralin (200°C), which can’t be explained within the scheme 24, as it is known, that perfluoroalkyl groups in aromatic ring of substrate hamper its reactions with electrophilic agents.
Besides that, in case of tetralin 32 according to Scheme 24 we shoudn’t have expected the forming of product 33, but indan 33a as the arenium ion 39 leading to the compound 33 must be less stable [C2F5 group is located in resonance (*) position] than ion 39a, while the stability of tetralinyl cations 40 and 40a is comparable (Scheme 26) [50].
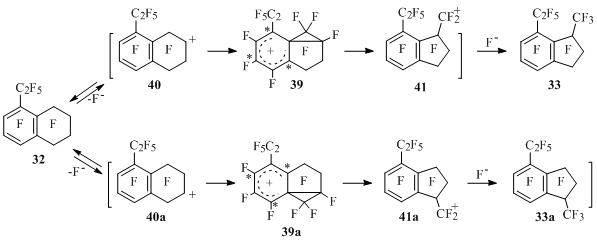
Scheme 26
In connection with that another way of reaction passing has been offered – the contraction of ring in intermediate tetralin-1-yl cations [50]. It is supposed, that the isomerization of tetralin-1-yl cations 42 and 42a into indanylmethyl ions 41 and 41a for example by 1,2-shift of perfluoroalkyl fragment through transition state 43 and 43a correspondingly (Scheme 27) will be a limiting stage for the process.
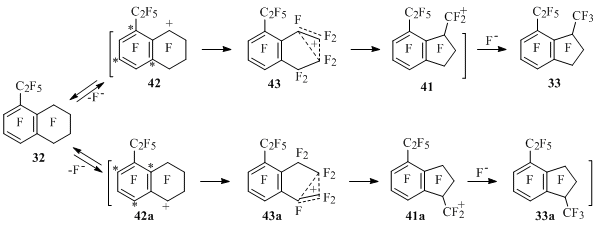
Scheme 27
Analyzing relative stability of intermediate cations 41, 41a, 42 and 42a we can draw a conclusion, that indan 33 must be mainly obtained (and is being obtained) from tetralin 32, and not its isomer 33a. Indeed, the energy of cation 42 is higher compare to ion 42a as in it the C2F5 group is located in resonance position. At the same time indanylmethyl ions 41 and 41a must differ from each other significantly less in energy terms. Thus, the energy barrier for transforming of cation 42 to cation 41 must be lower than for the isomerization of cation 42a into cation 41a. Smaller reactivity of perfluoroteralin compared to 2-halogenpolyfluoroteralins and ethyl and diethyltetralines 32 and 36 is explained analogously [50].
A considered way of process passing conforms with the fact, that at electrophilic chlorination of 2-methylnaphtalene perchloro-3-vinylindene [52] (Scheme 28) is obtained, and at chlorination of naphthalene-(1-14C) carbon atom located in position 2 of naphatlene is eliminated forming CCl4 and perchlorinated indan and indene [53].
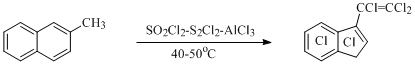
Scheme 28
Possible mechanism of indan 33to 34 isomerization and a way of contraction of five-membered ring to four-membered one in indan 37 forming compound 38 are presented at Scheme 29 [50]. Cations 44, in which the opening of five-membered ring forming cations 45 occurs must be generated from compounds 33 and 37. Intramolecular cyclization of ion 46 obtained from cation 45a and further attachment of fluorine anion to ion 47 produces compound 34.
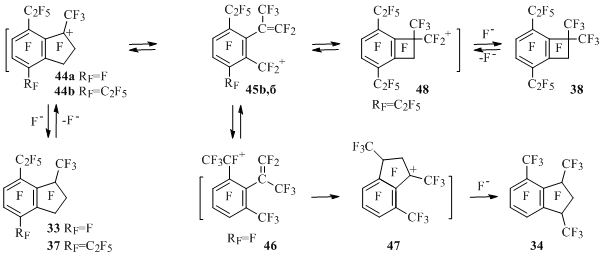
Scheme 29
Intramolecular attack of one or another vinyl atom of carbon by carbocation centre in ion 45b will lead to benzocyclobutene 38 or to the initial indan 37 after attachment of fluorine anion.
Skeletal transformations of other polyfluoroindanes also proceed through opening of five-membered cycle in indan-1-yl cations with intermediate formation of polyfluorostyrenes, which are closed intramolecularly forming new indanes or are fluorinated producing perfluoro-1,2-dialkylbenzenes. Thus, perfluoro-1,2-dialkylbenzenes are formed at heating of perfluorinated indan, 1-methyl-, 4- and 5-methylindanes with antimony pentafluoride and fluorination of aromatic ring takes place in perfluoro-2-methylindan [49] (Scheme 30).
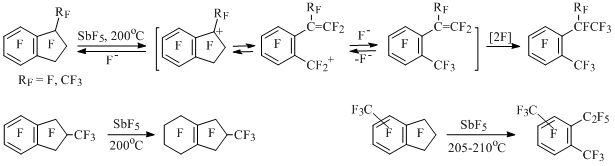
Scheme 30
In the reactions of perfluoro-1-isopropylindan (49) and perfluoro-1-ethylindan (50) with SbF5 the formation of polyfluorostyrenes is registered. Thus, isopropylindan 49 is isomerizing into tetramethylstyrene 51 under the influence of antimony pentafluoride at 90°C. Under more rigid conditions the last one is cyclizing into trimethylindan 52, which later looses CF4, producing dimethylindene 53. In absence of SbF5 the compounds 49, 51, 52 do not change at 130°C [54] (Scheme 31).
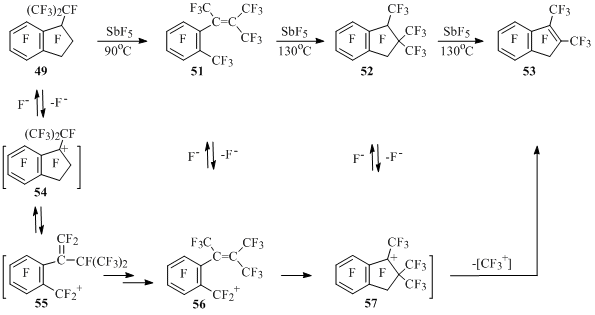
Scheme 31
It is supposed, that cation 54 is generated from indan 49 under the influence of SbF5, this cation is isomerizing into benzyl ion 55. The transition of double bond inside the chain forming styrene 51 occurs in the benzyl ion 55. Intramolecular cyclization of ion 56 generated from styrene 51 results in trimethylindanyl cation 57, from which at attachment of fluorine anion indan 52 is obtained, and at elimination of CF3+ – indene 53 [54] (Scheme 31).
At interaction of ethylindan 50 and antimony pentafluoride the products listed at Scheme 32 are formed, their ratio depends on the reaction conditions. It is stated, that compounds 53, 58, 60-63 can not be obtained from dimethylindan 59, as the indan 59 itself doesn’t form from these compounds in the medium of antimony pentafluoride.
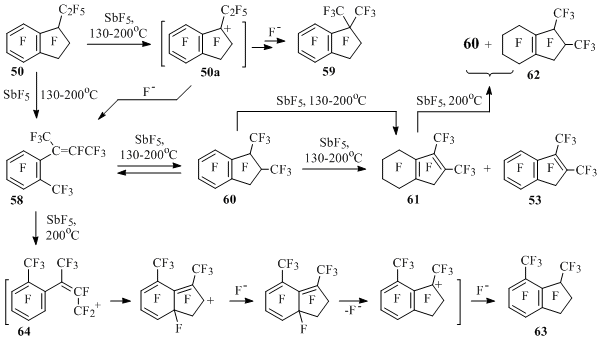
Scheme 32
Styrene 58 and indan 60 are transforming into each other in the presence of SbF5. Indan 60 along with disproportionation for indene 53 and diene 61 is being fluorinated to diene 61and later till product 62. Under the rigid conditions indan 63 is formed from compounds 58 and 60, this indan 63 can be obtained from diene 61 as well, as under the rigid conditions it can disproportionate into compounds 60 and 62 [55] (Scheme 32).
Isomerization of ethylindan 50 into dimethylindan 59 and styrene 58 must go with participation of perfluoro-1-ethylindan-1-yl cation (50a), in which on the one hand, 1,2-shift of trifluoromethyl group occurs forming indan 59 after attachment of fluoride anion, and on the other hand, - the opening of five-member cycle forming styrene 58 in the end. Indan 63 is obtained at high temperature probably as a result of intramolecular attack of the aromatic ring in cation 64 generated from styrene 58 [55] (Scheme 32).
Similar closure of propenyl fragment onto aromatic ring occurs at interaction of perfluorinated 1- and 3-phenylpropenes with aluminium chloride resulting in 1,1,3,3-tetrachlorohexafluoroindan and 1,1,3,3,5-pentafluoroindan [56].
At presence of cyclopropane ring in indan, which had been condense with five-member cycle the cyclopropane fragment is subject to opening under the influence of SbF5 with formation of perfluoro-1,1-dimethylindene or generation of perfluoro-1-methylindenyl cation [57] (Scheme 33).
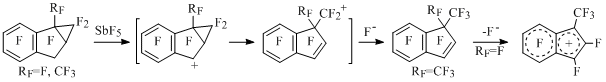
Scheme 33
The opening of three-membered cycle goes the same way as in perfluorinated alkylcyclopropanes under the influence of SbF5 [58] and aluminium chlorofluorides [59] with formation of perfluoroolefines.
In the reaction of perfluoro-1-methylbenzocyclobutene with AlCl3 and AlBr3 along with exchange of fluorine atoms by halogen the expansion of four-membered cycle up to five-membered one occurs resulting in 1,1,3,3-tetrahalogenpolyfluoroindanes [60,61]. At interaction of perfluorinated 1-methyl- (95°C) [60,61] and 1-ethylbenzocyclobutenes (130°C) [61] with antimony pentafluoride perfluorinated indan and 2-methylindan are being formed. Using the example of perfluoro-1-ethylbenzocyclobutene it is stated, that the reaction goes according to route 1 (Scheme 34) [61].
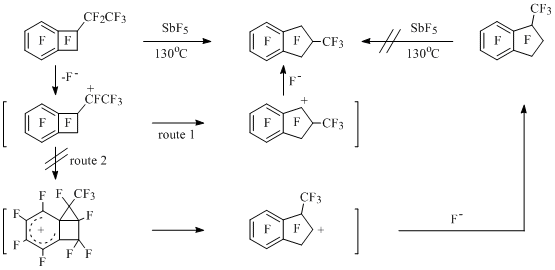
Scheme 34
As opposed to this one, in non-fluorinated series the expansion of four-member cycle is being carried out according to route 2. The present conclusion has been made based on results obtained during studying of acetolysis of tosilates of 1-(hydroxydideuteromethyl)benzocyclobutene [62].
The reaction of perfluoro-1-isopropylcyclobutene with SbF5 goes at 170°C, apparently, with participation of substituted isopropyl and benzocyclobutenyl cations in two directions with formation of products of four-membered cycle expansion and opening [54] (Scheme 35).
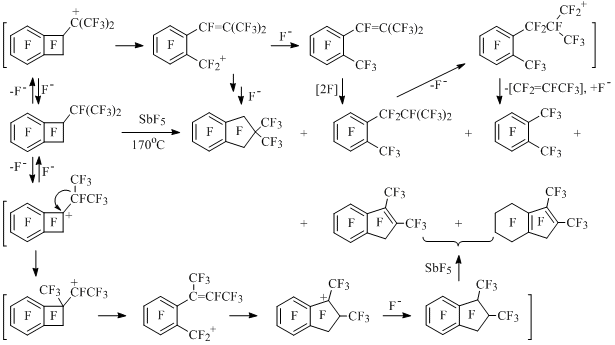
Scheme 35
The rearrangement of carbon framework in perfluorinated 1-methyl-1-isopropyl and 1- methyl-2-isopropylbenzocyclobutenes goes under the influence of antimony pentafluoride with participation of more stable methyl cation, at that it goes easier in 1,1-isomer, than in 1,2-isomer [54] (Scheme 36).
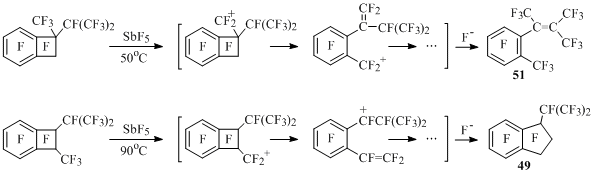
Scheme 36
Under more rigid conditions compounds 49 and 51 undergo further transformations [54] (Scheme 31). Similar regularities are observed in the reaction of 1,1- and 1,2-isomers of perfluoromethylethyl- and perfluorodiethylbenzocyclobutenes with antimony pentafluoride [63].
Perfluorobenzocyclobutene, which doesn’t possess a perfluoroalkyl group, dimerizes under the action of SbF5 producing 1-(2-ethylphenyl)benzocyclobutene (65). The reaction probably goes in a way of perfluorobenzocyclobuten-1-yl cation attacking the substrate molecule with further opening of four-membered cycle in σ-complex and catching the forming cation by fluorine anion [49] (Scheme 37).
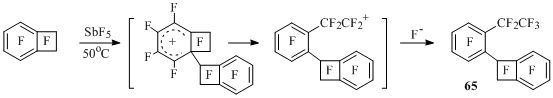
Scheme 37
In reactions of perfluorinated benzocyclobutene, 1-methyl- and 1-ethylbenzocyclobutenes with HF-SbF5 [64], Br2-SbF5 [49,65] and perfluorobenzocyclobutene with I2-SbF5 [66] the similar electrophilic opening of four-member cycle of substrate with formation of 2-X-perfluoroalkylbenzene also takes place (X=H,Br,I). Besides that, in the last reaction perfluoro-ortho-xylene [66] is formed, and corresponding perfluoro-ortho-dialkylbenzenes [66, 67] are obtained from perfluorinated 1-alkyl- and 1,2-diethylbenzocyclobutenes (Scheme 38).
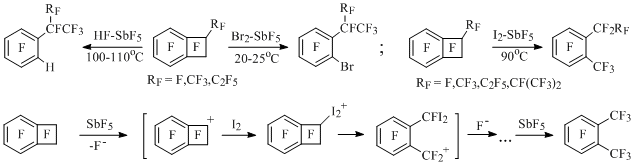
Scheme 38
At heating with antimony pentafluoride at 170°C perfluoro-1-phenylbenzocyclobutene returns with no alteration of carbon framework in the form of 1-hydroxyperfluoro-1-phenylbenzocyclobutene after hydrolysis of the reaction mass. As opposed to that, perfluorinated 1-(2-ethylphenyl)- (65) and 1-(4-ethylphenyl)benzocyclobutenes (66) under analogous conditions undergo skeletal transformations [31].
Thus, perfluoro-1,9-dimethyl-5,6,7,8-tetrahydrofluorene (67) is obtained from compound 65 along with derivatives of diphenylmethane 68 and 69, at that when reaction time is increased the content of product 67 in mixture increases, and the content of compounds 68 and 69 – falls (Scheme 39).
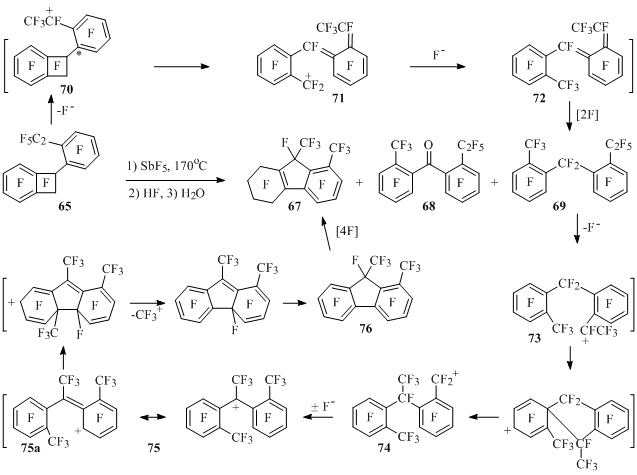
Scheme 39
The reaction must go as follows. Benzyl cation 70, in which a part of positive charge gets onto carbon atom (*) connected to benzocyclobutenyl fragment is being generated from compound 65 under the influence of antimony pentafluoride. In this cation as in similar cations from perfluoro-1-alkylbenzocyclobutenes the opening of four-member cycle occurs forming cation 71. Ion fluoride attachment to the last mentioned and further fluorination of compound 72lead to product 69.
It may be supposed, that further transformations of compound 69 go according to scheme 39including intramolecular rearrangement of cation 73 into ion 74, which isomerizes into cation 75. In the last mentioned the attack of positive charged carbon atom of one aromatic ring (resonance structure 75a) on another ring with CF3+ elimination after isomerization results in perfluoro-1,9-dimethylfluorene (76), which being fluorinated produces compound 67.
1-(4-Ethylphenyl)benzocyclobutene 66 at interaction with antimony pentafluoride as well as its isomer 65 at first produces the opening product of four-membered cycle 77. In cation 78 formed from it benzyl carbon atom attacks neighboring six-member ring, that in the end leads to derivatives of anthracene 79 and 80 [31] (Scheme 40).
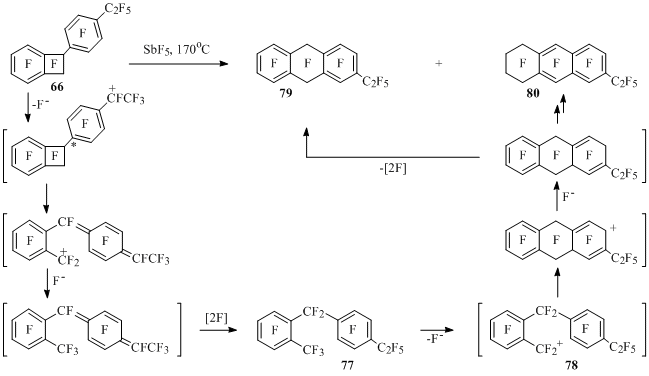
Scheme 40
At heating up the equimolar quantities of perfluoro-1-phenyl-1,2-diethylbenzocyclobutene and HF in excess of antimony pentafluoride along with opening of four-membered cycle typical for perfluoroalkylbenzocyclobutenes an unusual rearrangement of substrate carbon framework takes place including expansion of pentafluorobenzene ring to seven-membered cycle [68] (Scheme 41).
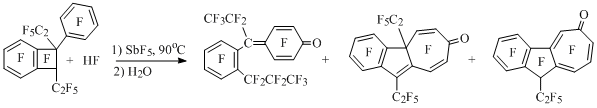
Scheme 41
At interaction of perfluoro-1-phenylindan (81) with antimony pentafluoride fluorene 83, octahydroanthracene 84 and compound 85 are formed, this compound is a product of contracnion of six-membered cycle to five-membered one in octahydroanthracene 84 [26] (Scheme 42).
In phenyltetralin 82 under the influence of SbF5 at first the contraction of alicyclic fragment takes place leading to methylphenylindene 86 and methylphenylindan 87, from which compounds 84 and 85 are formed later [69] (Scheme 42). The contraction of the ring in tetralin 82 and compound 84 is described by the scheme analogous to scheme 27 and the formation of products 83 and 84 – by the schemes similar to schemes 39 and 40.
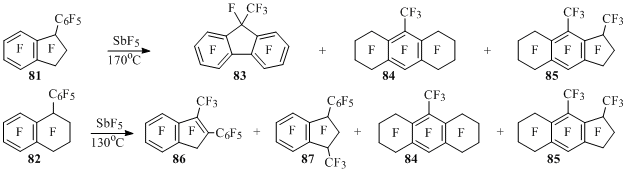
Scheme 42
It turned out, that carbonyl derivatives of perfluorobenzocycloalkenes are also subject to carbocationoid skeletal transformations under the influence of SbF5. At that methylphthalide 88 [41] is obtained from perfluoroindan-1,3-dione in the medium of antimony pentafluoride, and in the reaction with HF/SbF5 methylenephthalide 89 is formed, which at heating with SbF5 is being fluorinated to methylphthalide 88 [70] (Scheme 43).
Under the influence of SbF5 the decomposition of C(O)–C(O) bond occurs in perfluorobenzocyclobutendione, it leads to formation of tetrafluorophtalic acid after hydrolysis of the reaction mass [42]. Perfluoro-3-ethylphthalide is the main product in the reaction of perfluorotetralin-1,4-dione and antimony pentafluoride [42] (Scheme 43).
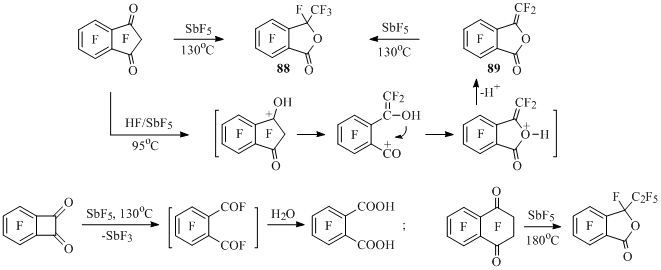
Scheme 43
At interaction of perfluorobenzocycloalken-1-ones and antimony pentafluoride along with skeletal transformations of ketones their disproportionation into benzocycloalkenes and benzocycloalkendiones takes place, these compounds later react forming the mixture of products [42] (Scheme 44).
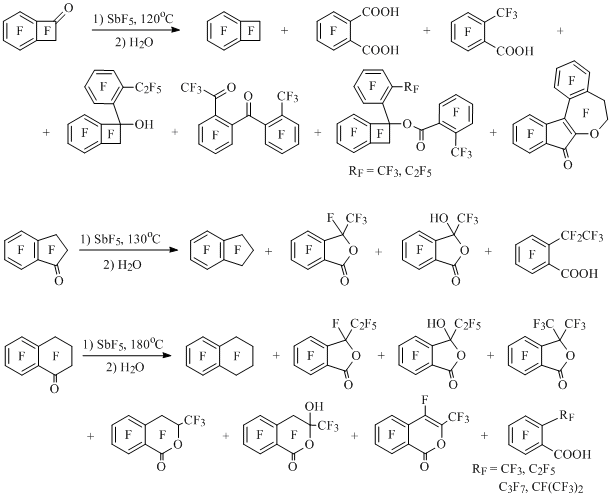
Scheme 44
Under the influence of SbF5 at 70°C in perfluoro-3-ethylindan-1-one (90) the opening of five-membered cycle occurs later resulting in formation of acid 91 after the hydrolysis of reaction mass. Under more rigid conditions the solution of salts of isochromenyl cations 92 and 93 is formed, during the hydrolysis of these cations the corresponding isochromen-1-ones are formed. At increasing the reaction period cation 92 is isomerized into thermodynamically more stable ion 93 [43] (Scheme 45).
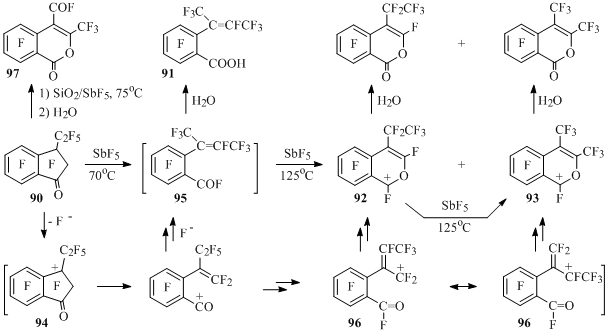
Scheme 45
The transformations of ketone 90 can occur with participation of cation 94, in which the five-membered cycle opens forming fluoroanhydride 95. Intramolecular cyclization of cation 96 with participation of one or another allylic system charge center and further transition of double bond inside the cycle result in formation of cations 92 and 93 [43] (Scheme 45).
As opposed to indanone 90 perfluoro-3,3-diethylindan-1-one doesn’t change under the influence of SbF5 even at 130°C. However, under more rigid conditions diethylindanone undergoes some skeletal transformations, opening mechanism of five-membered cycle in it differs [71] (Scheme 46) from cycle opening mechanism in ethylindanone 90 [43](Scheme 45).
Scheme 46
Indanone 90 reacts with SiO2 in the medium of SbF5 forming compound 97 after hydrolysis of the reaction mass [43] (Scheme 45). The same compound forms at interaction of perfluoro-1-ethylindan (50) with excess of SiO2 in the presence of SbF5 [43].
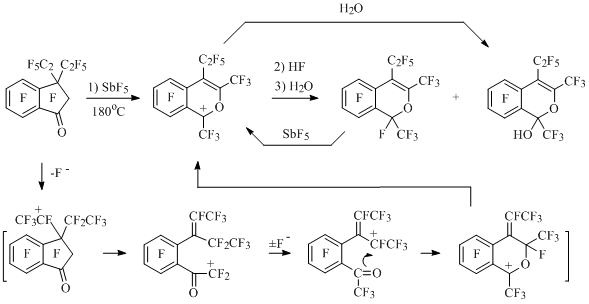
In the reaction of perfluoro-1-methylindan (98) with an excess of SiO2 in the medium of SbF5 6-(carboxymethyl)-2,3,4,5-tetrafluorobenzoic acid is obtained at 70°C after hydrolysis, and with lack of SiO2 – perfluoro-3-methylindan-1-one is obtained, from which perfluoro-4-methylisochromenyl cation (99) is formed (Scheme 47) [72] in the medium of SbF5.
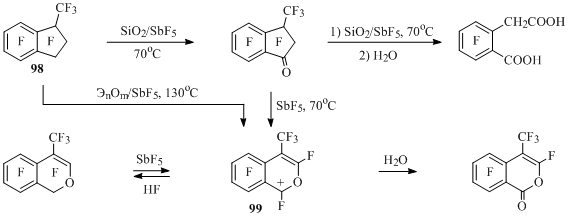
Scheme 47
At heating up of methylindan 98 with SbF5 in a glass tube at 130°C the solution of cation 99 salt is formed, which is a product of interaction of compound 98 and antimony pentafluoride and glass as a source of inorganic oxides (ЭnOm). At hydrolysis of a reaction mixture perfluoro-4-methylisochromen-1-one is obtained, and at quenching with anhydrous hydrogen fluoride – perfluoroperfluoro-4-methyl-1H-isochromene [72,73] (Scheme 47).
Cation 101 salt solution producing phthalide 102 at hydrolysis is formed from ethylphenylbenzocyclobutenone 100 and antimony pentafluoride at 70°C [74] (Scheme 48).
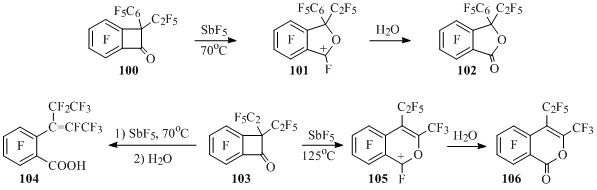
Scheme 48
At 70 °C under the influence of SbF5 the openining of four-membered cycle is taking place in diethylbenzocyclobutenone 103, which leads to the formation of acid 104 after the hydrolysis of the reaction mass. Under more rigid conditions the salt solution of isochromenyl cation 105 is being formed, at hydrolysis of which isochromenone 106 is obtained [74] (Scheme 48).
Thus, the studying of electrophilic reactions, carbocations and cationoid skeletal transformations of polyfluoroarenes and their derivatives is a fruitful direction of studies, during which the regularities of fundamental character have been found widening our knowledge on chemical behavior of aromatic compounds. The reactions considered proved to be perspective for the synthetics as well. Hard to obtain polyfluorinated derivatives of cyclohexadienes, cyclohexadienones, fluorenes and also perfluorinated ortho-dialkyl- and ortho-halogenalkylbenbenzenes, alkylphthalides, alkylisochromenones and their ketoderivatives were obtained with their help. It can be supposed, that further studying of cationoid reactions of polyfluoroarenes would lead to obtaining of new information on the mechanism of that reactions, development of new synthesis methods of various derivatives of polyfluoroarenes.
References
2. Nield E., Stephens R., Tatlow J.C. // J. Chem. Soc. 1959. N 1. P. 166-167.
3. Pummer W.J., Wall L.A. // J. Res. NBS. 1959. V. 63A. P. 167-169; Chem. Abstr.1960.V.54.10906h.
4.YakobsonG.G.,ShteingartsV.D.,VorozhcovN.N. // Zhurn.Vsesoyuz. khim. o-va im. D. I. Mendeleeva 1964. T. 9. N6. S. 702-704.
5. Shteingarts V.D., Yakobson G.G., Vorozhcov N.N. // Dokl. AN SSSR. 1966. T. 170. N6. S.1348-1351.
6. Shteingarts V.D., Yakobson G.G.// Zhurn. Vsesoyuz. khim. o-va im. D.I.Mendeleeva 1970.T. 15. N1. S. 72-80.
7. Shteingarts V.D., Kobrina L.S., Bil'kis I.I., Starichenko V.F. // Khimiya poliftorarenov: mekhanizm reakcij, intermediaty. Novosibirsk: Izd-vo «Nauka» Sib. Otd. 1991. 272 s.
8. Shteingarts V.D. // Uspekhi khimii. 1981. T. 50. N8. S. 1407-1436.
9. Shteingarts V.D. // Zhurn. organ. khimii. 1975. T. 11. Vyp. 2. S. 461.
10. Shteingarts V.D., Dobronravov P.N. // Zhurn. organ. khimii. 1976. T. 12. Vyp. 9. S. 2005-2012.
11. Dobronravov P.N., Shteingarts V.D. // Zhurn. organ. khimii. 1977. T. 13. Vyp. 8. S. 1679-1684.
12. Shteingarts V.D.,Pozdnyakovich Yu.V.,Yakobson G.G. //Chem. Commun. 1969. N21. P.1264-1265.
13. Dobronravov P.N., Shteingarts V.D. // Zhurn. organ. khimii. 1981. T. 17. Vyp. 10. S.2245-2246.
14. Karpov V.M., Platonov V.E., Yakobson G.G. // ZHurn. organ. khimii. 1974. T. 10. Vyp. 3.S. 663-664.
15. Karpov V.M., Platonov V.E., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1976. N11. S. 2647-2648.
16. Karpov V.M., Platonov V.E., Yakobson G.G. // Tetrahedron. 1978. V. 34. N21. P. 3215-3218.
17. Karpov V.M., Platonov V.E., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1979.N9.-S.2082-2087.
18. Karpov V.M., Platonov V.E., Shchegoleva L.N. // Zhurn. organ. khimii. 1998. T. 34. Vyp.11. S. 1732-1737.
19. Karpov V.M., Platonov V.E., Chujkov I.P., Shchegoleva L.N. // Zhurn. organ. khimii. 1999.T. 35. Vyp.1. S. 87-91.
20. Dewar M. // Teoriya molekulyarnyh orbitalej v organicheskoj khimii. M.:Mir. 1972. 592s.
21. Breslow R. // Chem. Eng. News. 1965. V. 43. N 26. P. 90-99.
22. Breslow R. // Pure Appl. Chem. 1971. V. 28. N 2-3. P. 111-130.
23. Breslow R. // Acc. Chem. Res. 1973. V. 6. N 12. P. 393-398.
24. Shchegoleva L.N., Karpov V.M., Platonov V.E. // Zhurn. organ. khimii. 2002. T. 38. Vyp. 7.S. 1037-1042.
25. Karpov V.M., Platonov V.E. // Zhurn. organ. khimii. 2001. T. 37. Vyp.7. S. 1033-1039.
26. Karpov V.M., Mezhenkova T.V., Platonov V.E., Sinyakov V.R. // J. Fluorine Chem. 2001. V. 107. N 1. P. 53-57.
27. Pozdnyakovich Yu.V., Shteingarts V.D. // Zhurn. organ. khimii. 1978. T. 14. Vyp. 3. S. 603-609.
28. Olah G.A., Comisarow M.B. // J. Amer. Chem. Soc. 1967. V. 89. N 4. P. 1027-1028.
29. Pozdnyakovich Yu.V. Shteingarts V.D. // J. Fluorine Chem. 1974. V. 4. N 3. P. 283-296.
30. Pozdnyakovich Yu.V. Shteingarts V.D. // J. Fluorine Chem. 1974. V. 4. N 3. P. 297-316.
31. Karpov V.M., Mezhenkova T.V., Platonov V.E., Sinyakov V.R. // J. Fluorine Chem. 2002.V. 117.N1.P. 73-81.
32. Karpov V.M., Platonov V.E. // Zhurn. organ. khimii. 1994. T. 30. Vyp. 5. S. 789.
33. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1985. N 10. S. 2315-2323.
34. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G. // J. Fluorine Chem. 1985. V. 28.N1.P. 121-137.
35. Karpov V.M., Mezhenkova T.V., Platonov V.E., Sinyakov V.R., Shchegoleva L.N. // Zhurn. organ.khimii.2002. T. 38. Vyp. 8. S. 1210-1217.
36. Sinyakov V.R., Mezhenkova T.V., Karpov V.M., Platonov V.E. // Zhurn. organ. khimii. 2007. T. 43. Vyp. 12. S. 1678-1686.
37. Sinyakov V.R., Mezhenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Yu.V. // Zhurn. organ. khimii. 2003. T. 39. Vyp. 6. S. 886-891.
38. Sinyakov V.R., Mezhenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., GatilovYU.V. // Zhurn. organ. khimii. 2006. T. 42. Vyp. 1. S. 85-93.
39. Pozdnyakovich Yu.V. Shteingarts V.D. // J. Fluorine Chem. 1974. V. 4. N 3. P. 317-326.
40. Karpov V.M., Panteleev I.V., Platonov V.E. // Zhurn. organ. khimii. 1991. T. 27. Vyp.10. S. 2183-2191.
41. Zonov Ya.V., Karpov V.M., Platonov V.E. // J. Fluorine Chem. 2005. V. 126. N 4. P. 437-443.
42. Zonov Ya.V., Karpov V.M., Platonov V.E., Gatilov Yu.V. // Zhurn. organ. khimii. 2008. T. 44. Vyp. 2. S. 212-226.
43. Zonov Ya.V., Karpov V.M., Platonov V.E. // J. Fluorine Chem. 2007. V. 128. N 9. P. 1065-1073.
44. Zonov Ya.V., Karpov V.M., Platonov V.E. // J. Fluorine Chem. 2007.V. 128.N9.P. 1058-1064.
45.KarpovV.M.,MezhenkovaT.V.,PlatonovV.E.,YakobsonG.G. //Zhurn.organ.khimii.1984.T. 20. Vyp. 6. S. 1341-1342.
46. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1987. N 6. S. 1361-1368.
47. Pozdnyakovich Yu.V., Bardin V.V, Shtark A.A, Shteingats V.D. // Zhurn. organ. khimii. 1979. T. 15. Vyp. 3. S. 656.
48. Bardin V.V., Furin G.G., Yakobson G.G. // J. Fluorine Chem. 1979. V. 14. N 6. P. 455-466
49. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G. // Bull. Soc.Chim.Fr.1986.N6.P. 980-985.
50. Karpov V.M., Mezhenkova T.V., Platonov V.E. // Zhurn. organ. khimii. 1997. T. 33. Vyp. 5. S. 755-761.
51. Karpov V.M., Mezhenkova T.V., Platonov V.E. // J. Fluorine Chem. 1996. V. 77. N 1. P. 101-103.
52. Garcia R., Riera J., Carilla J., Julia L., Molins E. // J. Org. Chem. 1992. V. 57. N 21. P. 5712-5719.
53. Zorkendorfer E., Boberg F. // J. Label.Comp. 1972.V. 8.N4.P. 567-574.
54. Karpov V.M., Mezhenkova T.V., Platonov V.E. // Izv.AN SSSR. Ser. khim. 1992. N 6. S. 1419-1423.
55. Karpov V.M., Mezhenkova T.V., Platonov V.E. // Izv. AN SSSR. Ser. khim. 1990. N3. S. 645-652.
56. Platonov V.E., Dvornikova K.V. // Izv. AN SSSR. Ser. khim. 1989. N 12. S. 2854-2857.
57. Karpov V.M., Platonov V.E. // Izv. AN SSSR. Ser. khim. 1998. N 8. S. 1666-1667.
58. Chepik S.D., Petrov V.A., Galahov M.V., Belen'kij G.G., Mysov E.I., German L.S. //Izv. AN SSSR, Ser. khim. 1990. N 8. S. 1844-1851.
59. Petrov V.A., Krespan C.G., Smart B.E. // J. Fluorine Chem. 1996.V. 77.N2.P. 139-142.
60. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G. // Izv.ANSSSR.Ser.khim. 1980.N5.S. 1212-1213.
61. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G. // J. Fluorine Chem. 1985.V. 28. N 1.P. 115-120.
62. Whitlock H.W., Fuch Ph. // Tetrahedron Lett. 1968. N 12. P. 1453-1456.
63. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1990. N 5. S. 1114-1120.
64. Karpov V.M., Mezhenkova T.V., Platonov V.E., Shchegoleva L.N. // Izv. AN SSSR. Ser. khim. 1991. N 11. S. 2618-2622.
65. Karpov V.M., Mezhenkova T.V., Platonov V.E. // J. Fluorine Chem. 1996. V. 77. N 2. P.133-137.
66. Karpov V.M., Mezhenkova T.V., Platonov V.E. // Mendeleev Commun.1997. N2. P. 71-72.
67. T.V. Mezhenkova, V.M. Karpov, V.E. Platonov. // ZHurn. organ. himii. 2010. T.46. Vyp. 9.S.1414-1416.
68. Mezhenkova T.V., Sinyakov V.R., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Yu.V. // J. Fluorine Chem. 2008. V. 129. N 1. P. 64-67.
69. Sinyakov V.R., Mezhenkova T.V., Karpov V.M., Platonov V.E. // J. Fluorine Chem. 2004.V. 125. N 1. P. 49-53.
70. Zonov Ya.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Yu. V. // J. Fluorine Chem. 2006.V. 127. N 12. P. 1574-1583.
71. Zonov Ya.V., Karpov V.M., Platonov V.E. // Zhurn. organ. khimii. 2011. T. 47. N 2. S. 217-222.
72. Zonov Ya.V., Mezhenkova T.V., Karpov V.M., Platonov V.E. // Zhurn. organ. khimii. 2008. T. 44. Vyp 11. S. 1675-1679.
73. Karpov V.M., Mezhenkova T.V., Platonov V.E., Yakobson G.G.// Izv. AN SSSR. Ser. him. 1991. N 3. S. 745-746.
74. Zonov Ya.V., Mezhenkova T.V., Karpov V.M., Platonov V.E. // J. Fluorine Chem. 2008. V. 129.N 12. P. 1206-1208.
Recommended for publication by Prof. V.E. Platonov
Fluorine Notes, 2011, 79, 1-2
