Received: July, 2011
Fluorine Notes, 2011, 79, 3-4
Features of phenyl ethers formation using polyfluoroalkyl chlorosulfites
Rakhimov A.I, Miroshnichenko A.V.
Volgograd State Technical University, Russia, 400131, Volgograd, Lenin Prosp., 28
e-mail: organic@vstu.ru
Abstract: Synthesis of phenyl ethers has been studied using reactions of polyfluoroalkyl chlorosulphites with sodium phenolate, phenols in the presence of bases and catalysed by N,N-dimethylformamide. Phenyl polyfluoroalkyl ethers have been prepared by the catalytic synthesis in 82.9% yield.
Keywords:phenol, phenolate, phenyl ethers, polyfluoroalkoxybenzenes, polyfluoroalkyl chlorosulfites, phenylpolyfluoroalkyl sulfite, N,N-dimethylformamide.
Previously polyfluoroalkyl phenyl ethers have been prepared by reactions of bromobenzene, p-bromotoluene with alcoholates of polyfluorinated alcohol in the presence of copper in pyridine [1]. The moderate yield (48.5 – 60%), the severe reaction conditions (110 – 120 °C, 22 h) and the toxic pyridine make it difficult to use this method.
Polyfluoroalkyl chlorosulfites (PFACS) are known to react with 1-adamantanol in the presence of sodium hydroxide in dioxane to form 1-(polyfluoroalkoxy)adamantane in yield to 76% [2].
The ethers have been obtained by the reactions of PFACS with saturated monohydric and aromatic-aliphatic alcohols catalyzed by N,N-dimethylformamide (DMF) [3-6].
In this work, we have compared the earlier known method for the synthesis of polyfluoroalkyl ethers of phenol [1] with the new reactions of PFACS with sodium phenolate, phenols in the presence of a base (NEt3, K2CO3) catalyzed by DMF.
Phenol was introduced in the reaction with PFACS together with equimolar amount of triethylamine (ТEA), which appears to react via a six-membered transition state, like that reported in [7].
A solution of PFACS was added in a preliminarily prepared phenol–TEA complex at -10 ÷ -5 °C, then the reaction mixture was allowed to stand for 5 to 6 hours at room temperature. Pentane, hexane, or chloroform were used as the solvent, because the triethylammonium chloride that formed is not soluble in them, it readily precipitates and can be easily removed from the reaction mixture. Polyfluoroalkyl ethers of phenol have been prepared by this method in 42.5 – 51.0% yields. Disadvantages of this method is the necessity to use big amounts of ТEA, this results in formation of triethylammonium chloride as a by-product. The formation of this salt occurs with heat liberation that facilitates side reactions and decreases the yield of the ether. In addition, triethylammonium chloride is an undesirable contamination of the reaction product, which is difficult to remove.
Another method that we used was the reaction of sodium or potassium phenolates with PFACS. However, in this case the yield of polyfluoroalkyl ethers of phenol was low (35%). The yield of the ether in the reaction of phenol with PFACS in the presence of potassium carbonate proved to be much lower (only 16.2%). This can be explained by the formation of an unsymmetrical sulfite – phenyl polyfluoroalkyl sulfite – under the reaction conditions, which undergoes some transformations, like unsymmetrical anhydrides of carboxylic and alkylsulfinic acids [8].
C6H5ONa + Cl-S(O)O-CH2Rf → C6H5O-S(O)O-CH2Rf + NaCl
C6H5OH + K2CO3 + Cl-S(O)O-CH2Rf → C6H5O-S(O)O-CH2Rf +KCl +KHCO3
Rf = (CF2CF2)nH, n = 1, 2.
When unsymmetrical sulfite is heated in the presence of a base, its disproportionation occurs to give diphenyl ether, alkyl(phenyl) sulfinates and symmetrical dialkyl sulfites, which can further lead to phenyl ethers.
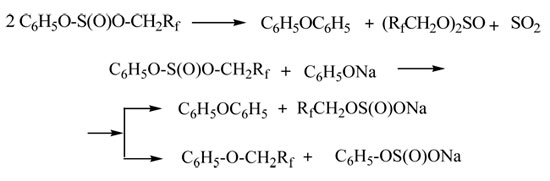
Sulfites and sulfinates can decompose in the presence of phenol to give the polyfluorinated alcohols.
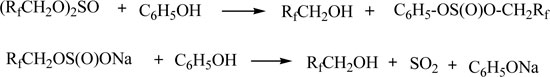
We showed that the new catalytic synthesis of polyfluoroalkyl ethers using the reaction of phenols with PFACS is most promising.
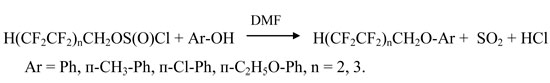
The reaction was carried out in the presence of a solvent (tetrachloromethane, chloroform, heptane, ether). The reactants were mixed at temperature –10 to –5 °C with polyfluoroalkyl chlorosulfite being dosed into a solution of phenol mixed with DMF at their molar ratio equal to (1 – 1.1) : 1 : 0.02, respectively. The reaction was carried out at 30 °C for 6 h. The gaseous reaction products liberated (sulfur dioxide and hydrogen chloride) were removed by dried air. The yields of ethers prepared by different methods and their physicochemical properties are given in the Table as follows.
Table. Polyfluoroalkyl phenyl ethers.
|
# |
Formula |
Yield, % |
bp, °C/mm Hg |
d204 |
nD20 |
|
1 |
C6H5OCH2CF2CF2H |
42.52 50.03 |
53/3 |
1.4378 |
1.4508 |
|
2 |
C6H5OCH2(CF2CF2)2H |
71.31 51.02 48.53 |
90/1 |
1.5760 |
1.4040 |
|
3 |
C6H5OCH2(CF2CF2)3H |
74.81 56.03 |
99/1 |
1.5550 |
1.3950 |
|
4 |
p-Me-C6H4-OCH2(CF2CF2)2H |
82.91 56.03 |
110/2 |
1.5280 |
1.4250 |
|
5 |
p-Me-C6H4-OCH2(CF2CF2)3H |
69.61 57.33 |
106/1 |
1.5440 |
1.3960 |
|
6 |
p-Cl-C6H4-OCH2(CF2CF2)2H |
45.01 |
108/1 |
1.6020 |
1.4390 |
|
7 |
p-EtO-C6H4-OCH2(CF2CF2)2H | 45.51 |
115/1 |
1.4136 |
1.4385 |
1– Catalytic synthesis in the presence of DMF, 2– synthesis in the presence of triethylamine, 3– synthesis from bromobenzene (toluene) in pyridine.
Like in the case of alcohols, in the reaction of PFACS with phenols catalyzed by DMF, nucleophilic substitution of chlorosulfite group by phenoxy group occurs with simultaneous liberation of hydrogen chloride and sulfur dioxide to form polyfluoroalkyl phenols.
Phenols having acidic properties associate with DMF to form donor-acceptor complexes (I), which are stable at low temperatures.
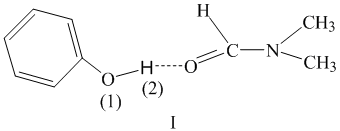
Because of formation of hydrogen bonds polarization and the H (2)- O(1) bond lengthening occur in associate (I) to increase proton mobility.
The first step of the reaction includes interaction of associate (I) with a PFACS molecule. In this case a six-membered complex (II) is formed as a result of interactions between the O(1)···C(3) and H(2)···Cl atoms. The high electron density at the chlorine atom and significant positive charge at the proton of the phenol hydroxy group facilitates the formation of this complex.
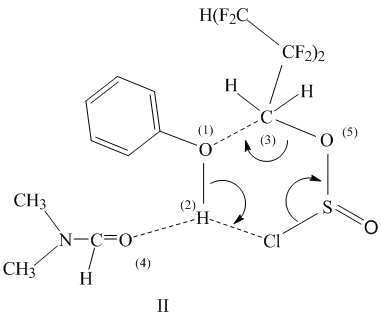
At the second step, the transfer of proton H(2) to the polar chlorine atom occurs in the complex and simultaneously the bonds C(3)-O(5) and S-Cl are cleaved with liberation of SO2 and HCl, and regeneration of DMF molecule with formation polyfluoroalkyl phenyl ether.
II → H(CF2CF2)nCH2O-C6H5 + SO2 + HCl + HC(O)-N(CH3)2
As was shown earlier by the example of benzyl alcohol [4], introduction of phenyl or other electron-acceptor substituent into an alcohol molecule increases proton mobility (acidity) at the expense of the higher polarization of the H(2) -O(1) bond; this results in an increase in reactivity of the alcohol in the reaction with polyfluoroalkyl chlorosulfite. On the other hand, introduction of electron-acceptor groups decreases the electron density at the oxygen atom and nucleophilicity of the alcohol, this makes it difficult its reaction with the carbon atom in polyfluoroalkyl chlorosulfite.
Kinetic studies show that in the case of alcohols, the proton transfer is the limiting factor that determines the formation of the reaction products [6].
The specific feature of the reaction of PFACS with phenols is the fact that phenols having more strong acidic properties than monohydric alcohols, are the weaker nucleophiles at the same time. Probably, it is the feature which determines their lower reactivity. It was shown that introduction of the strong electron-acceptor substituent (nitro group) decreases the reactivity of phenol to a such extent that preparation of polyfluoroalkyl ether of phenol fails.
Experimental
The IR spectra were recorded on a Spekord – M82 instrument, in thin layer (liquid films). 1H NMR spectra were recorded on a Mercury-300 (Varian) spectrometer operating at 300 MHz with tetramethylsilane used as an internal standard and carbon tetrachloride used as a solvent.
1. Synthesis of 1-(2,2,3,3–tetrafluoropropoxy)benzene.
C6H5OCH2CF2CF2H
Phenol (3.45 g, 0.04 mol) and triethylamine (3,71 g, 0.04 mol) were mixed in pentane (15 ml) until complete dissolution was achieved, the mixture was cooled to –10 °C and a solution of 1,1,3-trihydroperfluoropropyl chlorosulfite (7.87 g, 0.04 mol) in pentane (20 ml) was dosed with maintaining the above temperature. The reaction mixture was allowed to stand at room temperature for 24 h. Triethylamine hydrochloride was filtered off, pentane was distilled off and the following distillation under vacuum gave the title product. Yield 3.24 g, 42.5%, bp 53 ºC (3 mm Hg), nD20 1.4508, d204 1.4378. IR (ν, cm–1): 2932 (CH2), 1604, 1504 (Ph), 1239, 1032 (ν C-O-C), 1192 (νCF2).
2. Synthesis of 1-(2,2,3,3,4,4,5,5–octafluoropentoxy)benzene.
C6H5OCH2(CF2CF2)2H
Phenol (2,05 g, 0.0216 mol) was dissolved in chloroform (30 ml), mixed with DMF (0.034 ml, 0.0004 mol), cooled to –10 °C, and a solution of 1,1,5-trihydroperfluoropentyl chlorosulfite (6.80 g, 0.0216 mol) in chloroform (10 ml) with maintaining temperature –10 °C. After the reactants were mixed the temperature was increased to 30 – 35 °C and the reaction mixture was allowed to stand for 6 h with continuous dried air flush. The chloroform was distilled off and the product was distilled under vacuum to give the title compound (4,75 g) in 71,3 yield (bp 90 ºC /1 mm Hg, nD201.4040, d204 1.5760. IR (ν, cm–1): 1150s (νC-O-C); 1203s, 1256s (νCF2); 1317m, 1441m, 1485m; 1529s, 1635m, 1644m (Ph); 2897w, 2959m, 3012m (νCH2); 3065w (CHF2). 1H NMR (δ, ppm): 6.84 and 7.16 m (5H C6H5); 6.05 tt (53.0, 4.0) (1H, HCF2); 4.47 t (12) (2H, O-CH2-CF2).
3. Synthesis of 1-(2,2,3,3,4,4,5,5,6,6,7,7–dodecafluoroheptoxy)benzene.
C6H5OCH2(CF2CF2)3H
The title compound was prepared by the catalytic method in the presence of DMF using the procedure similar to that described in Example 2. Yield 74.8 %, bp 99 °C (1 mm Hg), nD20 1.3950, d204 1.5550. IR (ν, cm–1): 665s, 752s, 821s, 895m, 939m, 1000m, 1061s, 1113w, 1178s (νC-O-C); 1239s, 1283s (νCF2); 1452m, 1496m; 1530s, 1635s (Ph); 1704w, 1830w, 1915w, 1987w; 2896w, 2969m, 3013m (νCH2); 3065w, 3117w (CHF2).
4. Synthesis of 1-(2,2,3,3,4,4,5,5-octafluoropentoxy)-4-methylbenzene.
4-CH3-C6H4-OCH2(CF2CF2)2H
The title compound was prepared by the catalytic method in the presence of DMF using the procedure similar to that described in Example 2. Yield 82.9 %, bp 110 °C (2 mm Hg), nD201.4250, d204 1.5280. IR (ν, cm –1): 1150s (νC-O-C); 1203s, 1256s (νCF2); 1317m, 1441mр, 1485m; 1529s, 1635m, 1644m (Ph); 2897w, 2959m, 3012m (ν CH2, CH3); 3065w (CHF2). 1H NMR (δ, ppm, SSCC, J, Hz): 7.038 d (8.4) and 6.896 d (8.7) (4H, C6H4); 5.927 tt (51.6, 5.5) (1H, HCF2); 4.413 dq (108.5, 13.5) (2H, CH2); 2.229 (3H, CH3).
5. Synthesis of 1-(2,2,3,3,4,4,5,5,6,6,7,7-dodecafluoroheptyloxy)-4-methylbenzene.
4CH3-C6H4-OCH2(CF2CF2)3H
The title compound was prepared by the catalytic method in the presence of DMF using the procedure similar to that described in Example 2. Yield 69.6%, bp 106 °C (1 mm Hg), nD201.3960, d204 1.5440. IR (ν, cm–1): 1150s (νC-O-C); 1203s, 1256s (νCF2); 1317m, 1441m, 1485m; 1529s, 1635m, 1644m (Ph); 2897w, 2959m, 3012m (ν CH2, CH3); 3065w (CHF2). 1H NMR δ, ppm (SSCC, J, Гц): 7.052 m and 6.938 m (4H, C6H4); 5.935 tt (52.2, 5.4) (1H, HCF2); 4.421 dq (100.5, 13.2) (2H, CH2); 2.229 (3H, CH3).
6. Synthesis of 1-(2,2,3,3,4,4,5,5-octafluoropentoxy)-4-chlorobenzene.
4-Cl-C6H4-OCH2(CF2CF2)2H
The title compound was prepared by the catalytic method in the presence of DMF using the procedure similar to that described in Example 2. Yield 45.0 %, bp 108 °C (1 mm Hg), nD201.4390, d204 1.6020. IR (ν, cm–1): 613w, 639w, 682 and 710s, 770m, 830s, 878s, 939m, 990m, 1043s, 1122m, 1152s (νC-O-C); 1213s, 1262s (νCF2); 1317m, 1395w, 1430m, 1487m; 1513s, 1626m, 1687m (Ph); 1917w, 2052w; 2886w, 2930w, 2995m, 3039w (ν CH2, CH3); 3105w, 3135w (CHF2). 1H NMR (δ, ppm) (SSCC, J, Hz): 7.253 m and 7.059 m (4H, C6H4); 5.975 tt (52.2, 5.77) (1H, HCF2); 4.430 dq (97.8, 12.9) (2H, CH2).
7. Synthesis of 1-(2,2,3,3,4,4,5,5-octafluoropentoxy)-4-ethoxybenzene.
4-CH3CH2O-C6H4-OCH2(CF2CF2)2H
The title compound was prepared by the catalytic method in the presence of DMF using the procedure similar to that described in Example 2. Yield 45.5%, bp 115 °C (1 mm Hg), nD201.4385, d2041.4136. IR (ν, cm-1): 665m, 700w, 761s, 821m, 878m, 930, 982, 1000, 1061s, 1087s, 1178s (νC-O-C); 1213s, 1282s (νCF2); 1373m, 1452s, 1504m; 1540s 1556s 1643m, (Ph); 1917w, 2078w; 2930w, 2944w, 2978m, 3039s (ν CH2, CH3); 3100w, 3160w (CHF2).
References
- Sheludko, E.V. Zh.Org.Chem. / E.V. Sheludko, N. N. Kalibabchuk. - 1979. - vol. 15., Iss. 8. - p.1661-1665.
- Rakhimov, A.I. Zh.Org.Chem. / A. I. Rakhimov, O.V. Vostrikova. - 2002. - vol. 38., Iss. 7. - p. 1185-1188.
- Rakhimov, A.I. Zh.Org.Chem. / A. I. Rakhimov, A.V. Nalesnaya, O.V. Vostrikova - 2004. - vol. 40. , Iss. 4. - p. 967.
- Rakhimov, A.I., Zh. Obshch. Khim. / A. I. Rakhimov, R.V. Fisechko -2007, vol. 77, Iss. 10, p. 1750-1751.
- Rakhimov, A.I., Zh. Obshch. Khim. / A. I. Rakhimov, R.V. Fisechko -2008, vol. 78, Iss. 2, p. 338.
- Rakhimov, A.I., Zh. Obshch. Khim. / A. I. Rakhimov, A.V. Nalesnaya, R.V. Fisechko -2008, vol. 78, Iss. 101, p. 1842-1848.
- Rakhimov, A.I., Zh. Obshch. Khim. / A. I. Rakhimov, A.V. Nalesnaya, O.V. Vostrikova. - 2003. - vol. 39., Iss. 6- p. .949.
- Shigeru Oae, ORGANIC CHEMISTRY OF SULFUR / Springer; 1 edition (June 1, 1977)- 713 p.
Recommended for publication by Prof. Alexander I. Rakhimov
Fluorine Notes, 2011, 79, 3-4
