Fluorine Notes, 2011, 77, 1-2
СОЗДАНИЕ ОСНОВ И РАЗВИТИЕ ХИМИИ ПОЛИФТОРАРЕНОВ И –ГЕТАРЕНОВ
В.Е. Платонов, Т.Д. Петрова
Учреждение Российской академии наук Новосибирский институт органической химии им. Н.Н. Ворожцова Сибирского
отделения РАН (НИОХ СО РАН), Россия , г. Новосибирск, пр. Акад. Лаврентьева 9,
e-mail: petrova@nioch.nsc.ru
Аннотация. Обобщён ряд результатов пятидесятилетней деятельности лаборатории галоидных соединений НИОХ СО РАН по химии полифтораренов и –гетаренов. Эти результаты включают разработку методов синтеза полифтораренов и -гетаренов, с помощью которых получены собственно полифторарены и полифторхлорарены путём нуклеофильного замещения атомов хлора на фтор в перхлораренах при взаимодействии с фторидами щелочных металлов в отсутствие растворителя, получение из перфтор- и полифторхлораренов не полностью фторированных аренов путём гидрогенолиза связи С-F и C-Cl действием Zn(Cu) в водном ДМФА, а также методы функционализации перфтораренов в реакциях с источниками дигалокарбенов либо путём превращения уже имеющихся в молекуле функциональных групп при действии нуклеофильных, электрофильных и радикальных реагентов. Библиография – 95 ссылок.
I. Введение
Основным научным направлением лаборатории галоидных соединений с момента её основания и до настоящего времени было и остаётся исследование химии полифтораренов и –гетаренов. Интерес к этому типу соединений был вызван успехами их химии и выявившимися уникальными свойствами полифторорганических соединений других классов. Это стимулировало развитие методов синтеза и исследование реакционной способности полифтораренов и –гетаренов, которые до начала 60-х годов прошлого века были ещё мало известны, но, тем не менее, также могли оказаться весьма перспективными. Удобный метод получения первого представителя этого класса - гексафторбензола - был разработан в лаборатории галоидных соединений НИОХ СО АН и опубликован в 1963г [1]. Этот метод стимулировал бурное развитие химии полифтораренов и –гетаренов как в нашей стране, так и за рубежом, причём НИОХ и до настоящего времени является ведущей организацией в этой области. В лаборатории галоидных соединений разработка методов получения и исследование химии полифторароматических соединений обычно тесно переплетены, однако первоначально усилия были направлены специально на разработку удобного и эффективного метода синтеза ключевых полифторароматических соединений, чтобы сделать их доступными и получить возможность изучения их реакционной способности и областей применения. Изучение реакций этих соединений в различных условиях (термолиз, фотолиз), с реагентами различной природы (нуклеофилы, электрофилы, радикалы, карбены) неизбежно привело не только к накоплению знаний о реакционной способности полифтораренов и –гетаренов, но и к выявлению новых подходов к синтезу этих соединений. Эта работа проводилась на протяжении 50-ти лет существования лаборатории большим коллективом сотрудников, и большая часть основных результатов её отражена, помимо данной статьи, в других обзорных статьях, которые будут опубликованы вслед за данным сообщением. В настоящем обзоре рассмотрены разработанные в лаборатории методы синтеза полифтораренов и –гетаренов, которые можно разделить на три группы. Первая из них включает получение собственно перфтораренов и полифторхлораренов путём нуклеофильного замещения атомов хлора на фтор в перхлораренах под действием фторидов щелочных металлов в отсутствие растворителя. Вторая группа, позволяющая получать не полностью фторированные арены из перфтор- и хлорперфтораренов, основана на реакции гидрогенолиза последних, т.е. замещении атомов фтора и хлора на водород под действием Zn (Cu) в водном ДМФА. Наконец, третья группа включает методы функционализации перфтораренов действием дигалокарбенов, а также превращения уже имеющихся в молекуле полифторарена функциональных групп под действием нуклеофильных, электрофильных и радикальных агентов, что позволяет получать широкий набор соединений самых разных классов, включая гетероциклические производные. Круг превращений, входящих в эту группу, весьма обширен, и в настоящем обзоре рассмотрена только часть исследований, осуществлённых лабораторией в этом направлении.
II. Термолитические превращения
1. Нуклеофильные реакции. Введение атомов фтора в ароматическое кольцо реакцией нуклеофильного замещения хлора в перхлораренах действием фторидов щелочных металлов в отсутствие растворителя. Синтез перфтор- и полифтораренов.
В лаборатории галоидных соединений впервые в мире был разработан наиболее удобный и эффективный метод получения полифторированных ароматических соединений, который не утратил своего значения и сегодня. Он основан на реакции нуклеофильного замещения атомов хлора перхлорированных аренов на фтор под действием фторидов калия или цезия в отсутствие растворителя и при высокой температуре [1]. Создание его основывалось на более ранних работах ряда авторов этого метода по обмену атомов хлора на фтор в хлорнитробензолах под действием фторидов щелочных металлов в отсутствие растворителя [2,3]. Протеканию этих реакций способствовали активация атомов хлора нитрогруппой и применение повышенных температур. При реакции гексахлорбензола с фторидом калия в автоклаве при температуре 460-470°С [1] был получен продукт полного обмена - гексафторбензол с выходом 20-22%, и образовывалось большое количество продуктов неполного обмена, наиболее важным из которых являлся хлорпентафторбензол (24%). При увеличении температуры реакции до 530°С получалась смесь гексафторбензола и хлорполифторбензолов с выходом до 70%, а содержание гексафторбензола в смеси возрастало до 45%. Ректификацией смесь разделялась на компоненты. При этом гексафторбензол, хлорпентафторбензол и фракции соединений с меньшей степенью обмена хлора на фтор выделялись с выходами, делающими эти соединения доступными как в качестве объектов исследования, так и для промышленного использования [1,4]. Из фракции трихлортрифторбензолов выделен с выходом 60% твёрдый 1,3,5-трихлортрифторбензол.
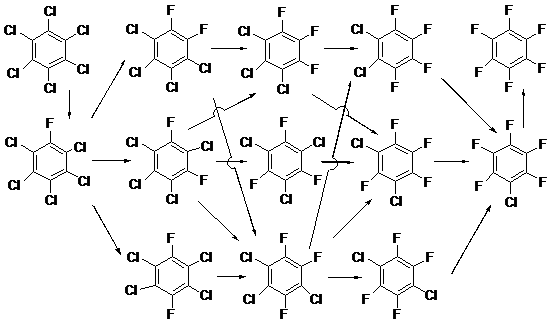
Обмен атомов хлора на фтор в перхлораренах под действием фторида калия в отсутствие растворителя оказался общим методом получения перфторароматических соединений из перхлорированных аналогов. Таким путём были синтезированы октафторнафталин [5], декафтордифенил [5], декафторпирен [6].
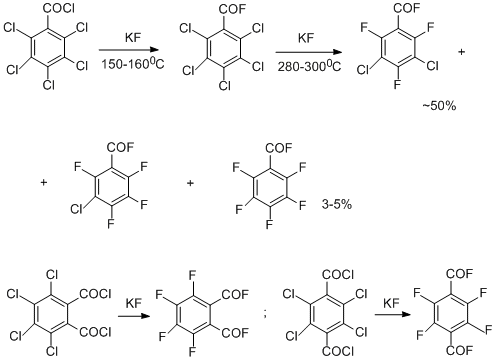
Метод оказался применимым и для получения некоторых функциональных производных полифтораренов, в частности, фторангидридов полифторароматических кислот. При этом реакция обмена, естественно, протекала в более мягких условиях вследствие дополнительного активирующего эффекта электроноакцепторных карбонильных группировок [7-9]. Обмен хлора на фтор был осуществлён и в полихлорированных хинонах. Именно так с 92%-ным выходом был синтезирован 1,2,3,4-тетрафторантрахинон из тетрахлорпроизводного [10]. Метод оказался применим даже к не полностью хлорированным неактивированным аренам, вплоть до хлорбензола, но при этом существенным фактором оказалось снижение термической стабильности как исходных, так и конечных соединений, содержащих атомы водорода, обуславливающее уменьшение выходов продуктов обмена, а также протекание более сложных превращений с образованием изомеров и продуктов восстановительного дегалоидирования [11].
С момента создания термического метода синтеза полифтораренов реакцией обмена хлора на фтор он широко используется во всём мире (Halex process) для получения соединений различных типов, включая широкий набор полифторированных гетероциклических систем [12]. Для синтеза гексафторбензола он до сих пор является лучшим и применяется в промышленном производстве этого соединения, хотя за длительное время своего существования усилиями различных исследовательских центров и фирм он модифицировался и совершенствовался, и это продолжается до сих пор в направлении поиска эффективных катализаторов межфазового переноса, приготовления фторида калия с высокоразвитой поверхностью, снижения температуры реакции и т.д. [13].
Следует отметить, что при детальном изучении реакционной смеси, полученной при взаимодействии гексахлорбензола с фторидом калия при высоких температурах и в отсутствие растворителя, в ней были идентифицированы в небольшом количестве перфторированные гомологи бензола – толуол, изомерные ксилолы, мезитилен, а также перфториндан. Предположение, что образование этих соединений может быть связано с участием в процессе дифторкарбена [14,15], стимулировало цикл работ по реакциям перфтораренов и их производных с дигалокарбенами.
2. Реакции с источниками дигалокарбенов
2.1. Реакции с источниками дифторкарбена. Трифторметилирование, циклоалкенилирование, полифторвинилирование полифтораренов, синтез полифторированных гетероциклических соединений
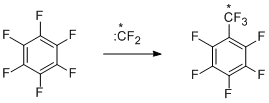
Для выявления наиболее вероятных путей образования перфторированных трифторметильных производных в реакции гексахлорбензола и КF (см. выше) изучено взаимодействие гексафторбензола с КF в аналогичных условиях [16] и установлено, что продуктами реакции являются те же трифторметильные производные, что и в случае гексахлорбензола. По-видимому, на первой стадии идёт взаимодействие гексафторбензола с КF и образование σ-комплекса, который распадается с выделением дифторкарбена. Последний в свою очередь взаимодействует с исходным гексафторбензолом, давая октафтортолуол и далее по аналогичной схеме другие продукты трифторметилирования [16].
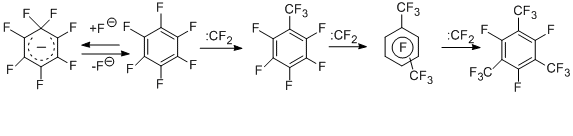
Карбенная схема образования трифторметильных производных подтверждается и результатами, полученными в реакциях перфтораренов с заведомыми источниками дифторкарбена – фторопластом-4 и тетрафторэтиленом. Данные для гексафторбензола [17,18] представлены на схеме ниже.
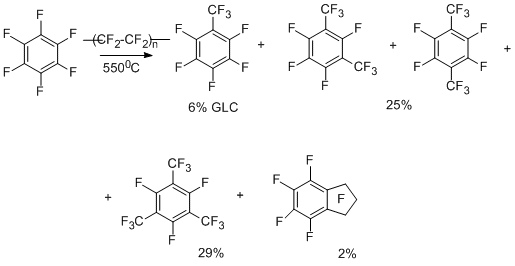
Проведение реакции с тетрафторэтиленом при более высокой температуре (720-770°С) [19] увеличивает степень превращения и выход продуктов (октафтортолуол 65-67%). В реакции трифторметилирования вступают также пентафторпиридин [20], декафториндан [21], декафтордифенил [21] и октафторнафталин [21].
Существенно, что при образовании политрифторметильных производных, как правило, наблюдается мета-ориентация, что согласуется с электрофильным характером дифторкарбена в указанных превращениях. Преимущественное образование в случае октафторнафталина 2-трифторметильного производного вместо более ожидаемого для электрофильных реагентов 1-изомера может быть обусловлено влиянием трифторметилирование термодинамических факторов, в результате чего более термодинамически устойчивый 2-изомер накапливается в смеси. Не исключается и стерическая затруднённость атаки дифторкарбеном α-положения октафторнафталина [19]. Процесс трифторметилирования является обратимым, и эта обратимость позволяет более обоснованно рассматривать влияние термодинамических факторов на реакцию трифторметилирования. Так, при нагревании октафтортолуола происходит его диспропорционирование с образованием гексафторбензола и перфторксилолов (с преобладанием мета-изомера [22]), изомерные ксилолы диспропорционируют на октафтортолуол и перфтормезитилен и при этом ещё наблюдается их "взаимопревращение" [23].
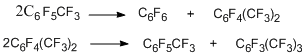
Закономерности, наблюдаемые в процессах диспропорционирования, позволяют сделать вывод, что и за их протекание может быть ответственен электрофильный дифторкарбен. Сумма данных, полученных в настоящее время по реакциям трифторметилирования, в особенности результаты реакции образования октафтортолуола из гексафторбензола и источника меченого дифторкарбена, в качестве которого использовался фтороформ, обогащённый изотопом 13С на 46% [24], позволяет предположить, что вероятным путём образования трифторметильных производных является внедрение дифторкарбена по связи С-F.
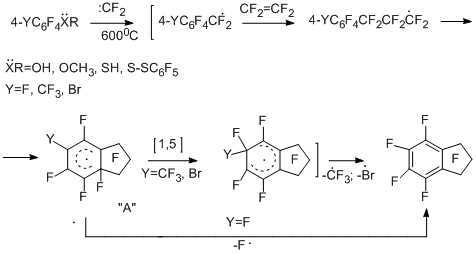
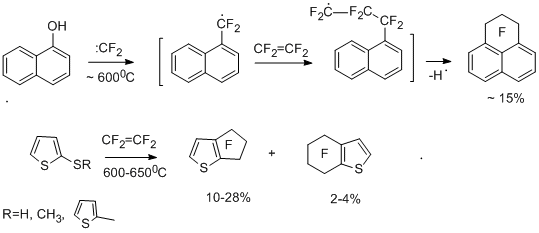
Введение в перфторароматическое соединение функциональных групп оказывает влияние на поведение полифторарена при взаимодействии с источником дифторкарбена. В случае электронодонорных заместителей реакционная способность возрастает, но взаимодействие идёт в основном по функциональной группе. При введении электроноакцепторных заместителей реакционная способность понижается, и в ряде случаев меняется направление реакции. В случае хлорпентафторбензола и тетрафторэтилена образовывались продукты, отвечающие внедрению дифторкарбена по связям С-Сl и C-F - гептафторбензилхлорид в смеси с хлоргептафтортолуолами наряду с перфтортрифторметильными производными и перфторинданом, в то время как бром- и иодпентафторбензолы реагируют более однозначно, давая в основном перфториндан [19]. 3-Хлортетрафторпиридин и 5-хлортетрафторпиримидин с тетрафторэтиленом при 820°С дают трифторметильные производные перфторпиридина и –пиримидина [19]. Реакции полифторированных бензольных производных, содержащих в функциональных группах атомы с неподелёнными парами электронов, с тетрафторэтиленом и другими источниками дифторкарбена протекают при более низкой температуре, и главным продуктом является перфториндан [25,26]. Интересно, что перфториндан образуется и при сопиролизе с тетрафторэтиленом перфтор-п-крезола [26], п-бромтетрафторанизола [19] и перфтор-п-тиокрезола [26], что может быть связано с изомеризацией радикального s-комплекса "А" за счёт 1,5- сдвига атома фтора и более лёгкого элиминирования радикалов CF3· и Br · из комплекса за счёт более низкой энергии связи С-С и С-Br по сравнению с С-F.
Представляется вероятным участие в образовании перфториндана перфторбензильных радикалов, реагирующих далее с тетрафторэтиленом с последующей внутримолекулярной циклизацией радикального типа. Действительно, гептафторбензилбромид, как заведомый источник гептафторбензильного радикала, при сопиролизе с тетрафторэтиленом в аналогичных условиях с высоким выходом даёт перфториндан [26]. В пользу предположения об образовании гептафторбензильного радикала свидетельствуют и данные по сопиролизу пентафторфенола с тетрафторэтиленом в присутствии брома или 1,2-дибромтетрафторэтаном, который в условиях реакции способен дебромироваться с образованием тетрафторэтилена. В результате этих реакций получены гептафторбензилбромид и перфторбензилфениловый эфир [27].
Возможный путь образования гептафторбензильного радикала на примере реакции пентафторфенола представлен ниже на упрощённой схеме. Он предполагает внедрение дифторкарбена по связи С-О [26].
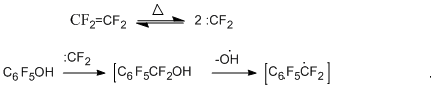
Описанный выше процесс полифторциклоалкенилирования с участием радикалов α,α-дифторбензильного типа в ароматическом ряду носит достаточно общий характер. Так, 4-метокситетрафторпиридин при сопиролизе с тетрафторэтиленом в качестве основного продукта даёт перфтор-2-пириндан [28].
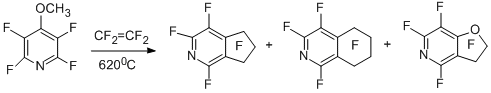
Полифторциклоалкенилирование имеет место и в реакциях нефторированных ароматических окси- и серасодержащих производных [29-32].
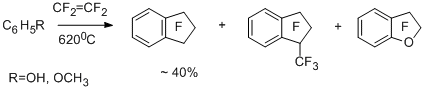
В случае пентафтортолуола и толуола реакции с тетрафторэтиленом идут в основном с участием пентафторбензильного и бензильного радикалов, приводя к 1,1-дигидрооктафториндану и 1,1-дигидро-2,2,3,3-тетрафториндану [33,34]. Более электронодонорный 2-метилтиофен образует в основном 4,4,5,5,6,6-гексафторциклопента[b]тиофен вследствие участия 2-тиенилдифторметильного радикала [34,35].
Закономерности реакций полифтораренов, имеющих электроноакцепторные хлоралкильные группы, и источников дифторкарбена отличаются от вышеописанных. Результатом реакций является образование продуктов фторвинилирования. Так, сопиролиз пентафторбензотрихлорида или его пара-замещённых производных с тетрафторэтиленом или хлордифторметаном приводит к полифторированным стиролам [36-38]. Процесс протекает путём внедрения дифторкарбена по связи С-Сl в бензильном положении с последующим дехлорированием или дегидрохлорированием промежуточных полифторхлорэтильных производных [36-38].
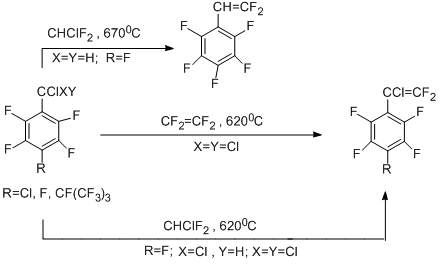
2.2. Реакции с источниками дихлоркарбена и пентафторбензотрихлоридом. Синтез полифтораренов с имидоилхлоридной группировкой
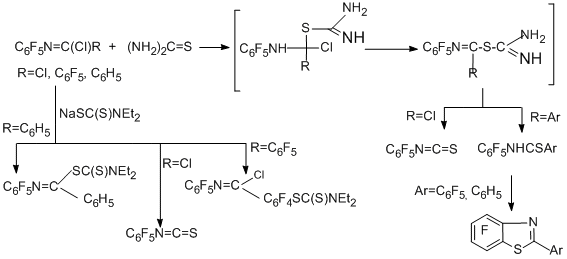
Термолитические реакции полифтораренов с источниками дихлоркарбена исследованы на примере полифторароматических аминов. Источником дихлоркарбена являлся хлороформ. В реакцию был введён также 2,3,4,5,6-пентафторбензотрихлорид как потенциальный источник пентафторфенилхлоркарбена. Сопиролиз полифторароматических аминов в проточной системе с этими соединениями при температурах 500-670°С протекает с участием только аминогруппы и не затрагивает полифторароматическое кольцо. В результате основными продуктами превращения с хлороформом являются N-полифторарилкарбонимидоилдихлориды, выход которых составляет 14-34% [39,40], а в реакции с пентафторбензотрихлоридом получаются N-(полифторарил)пентафторбензимидоилхлориды с выходом 50-77% [41].
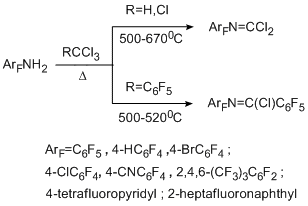
В реакции с 4-бромтетрафторанилином идёт обмен атома брома на хлор, и конечным продуктом оказывается 4-хлоримидоилхлоридное производное. Попытка вовлечь в превращение нефторированные амины, например, анилин не увенчалась успехом, по-видимому, вследствие термической нестабильности последнего. В то же время для получения полифторарилимидоилхлоридных производных метод оказался достаточно общим и применим не только к полифторированным аминам бензольного ряда, но также ряда нафталина и пиридина.
Возможные пути образования имидоилхлоридных производных на примере реакций с хлороформом представлены на схеме:
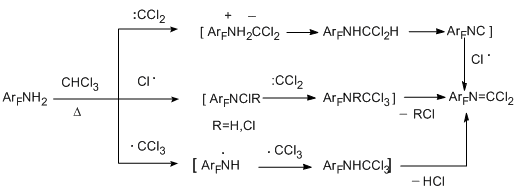
В случае хлороформа нельзя исключать и путь реакции с участием трихлорметильного радикала, хотя основное направление термического распада хлороформа идёт с образованием дихлоркарбена. Действительно, реакция с четырёххлористым углеродом, как заведомымм источником трихлорметильного радикала, приводит к тем же карбонимидоилдихлоридным производным и даже с несколько более высоким выходом (21-37%) за счёт уменьшения содержания побочных продуктов в реакционной смеси [40]. Реакция с пентафторбензотрихлоридом вероятнее всего также протекает по радикальному механизму.
3. Радикальные реакции
Термолитические радикальные превращения ароматических соединений и их полифторированных аналогов оказались перспективными для синтеза полифторированных гетероциклических соединений. Они базируются на идее образования из исходных аренов ArXR и ArFXR, содержащих функциональную группу с гетероатомом в бензильном положении (Х – гетероатом), радикалов бензильного ArX· и ArFX· или арильного Ar· и ArF· типа, их дальнейшем взаимодействии с полифторолефинами и последующей внутримолекулярной циклизации с образованием гетероциклической системы.
3.1. Реакции с участием бензолтиильных радикалов. Сопиролиз арилтиолов с полифторированными олефинами в присутствии окислителей. Синтез полифторированных дигидробензотиофенов
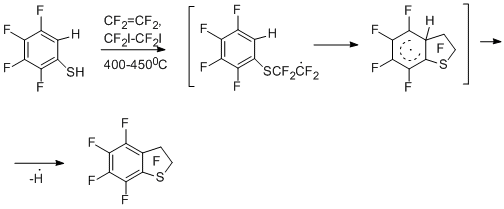
Примером реакций такого типа служит сопиролиз арилтиолов или их полифторированных аналогов с тетрафторэтиленом или хлортрифторэтиленом в присутствии окислителей. В качестве субстратов исследованы бензолтиол, дифенилдисульфид и их производные, легко генерирующие ArS· или ArFS· радикалы под действием окислителей (иод или его источник 1,2-дииодтетрафторэтан).
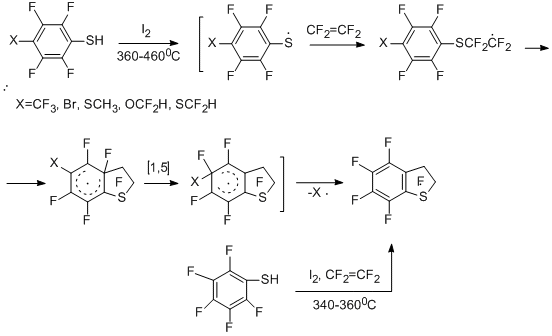
Продуктами реакций являются полифторированные 2,3-дигидробензо[b]тиофены [42,43]. Элиминирование заместителей в пара-замещённых полифторбензолтиолах [43] идёт по аналогии с реакциями этих соединений и тетрафторэтилена в отсутствие окислителя (см. раздел II.2.1). Сопиролиз 2-Н-3,4,5,6-тетрафторбензолтиола с тетрафторэтиленом в присутствии иода или 1,2- дииодтетрафторэтана даёт главным образом перфтор-2,3-дигидробензо[b]тиофен.
3.2. Реакции с участием радикалов фенильного типа
3.2.1 Сопиролиз полифторароматических нитро- и сульфонилхлоридных производных с тетрафторэтиленом. Образование перфтортетралина, -индана и их гетероаналогов.
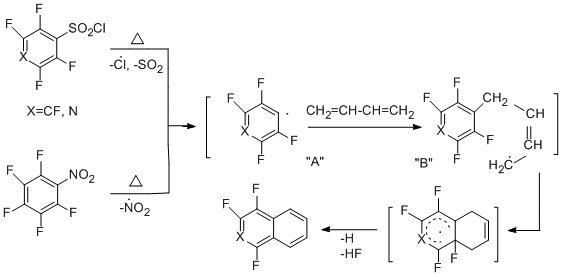
Весьма своеобразными оказались результаты сопиролиза тетрафторэтилена с пентафторнитробензолом, 4-нитротетрафторпиридином и пентафторбензолсульфонилхлоридом. В отличие от аналогичных реакций других производных полифтораренов с тетрафторэтиленом как источником дифторкарбена, описанных выше, в данном случае основными продуктами превращений оказались перфтортетралин и перфтор-5,6,7,8-тетрагидроизохинолин [19,25,28] наряду с перфторинданом и его гетероаналогом.
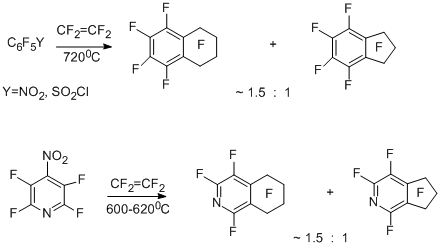
Такое направление реакции объясняется тем, что исходные нитро- и сульфонилхлоридные производные в условиях реакции генерируют полифторированные арильные радикалы, которые реагируют с молекулой тетрафторэтилена, давая радикалы перфторарилэтильного и затем перфторарилбутильного типа с последующей внутримолекулярной циклизацией последних, приводящей к образованию перфторированного шестичленного кольца.
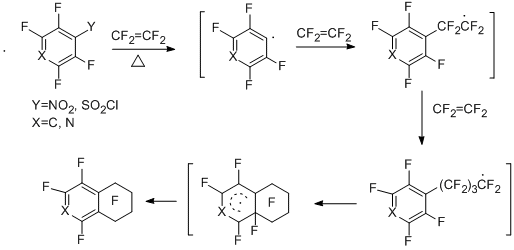
3.2.2. Сопиролиз полифторароматических нитро- и сульфонилхлоридных производных с бутадиеном. Образование 1,2,3,4-тетрафторнафталина и 1,3,4-трифторизохинолина
Сопиролиз пентафторбензолсульфонилхлорида, 2,3,5,6-тетрафторпиридин-4-сульфонилхлорида, а также пентафторнитробензола с бутадиеном приводит к 1,2,3,4-тетрафторнафталину в случае бензольных производных [44] и 1,3,4-трифторизохинолину в случае пиридинового производного [45].
Выход тетрафторнафталина из сульфонилхлоридного производного составляет 40%, из нитро – 20%, трифторизохинолин по данным ГЖХ получается с выходом 21%. Схема образования соединений включает термическую генерацию из исходных соединений полифторарильных радикалов типа "А", их взаимодействие с бутадиеном и образование аллильного радикала "В", его внутримолекулярную циклизацию и превращение образующегося радикального σ-комплекса в конечные продукты.
3.2.3. Cопиролиз полифторарентиолов с хлором или бромом. Получение хлор- и бромполифторбензолов
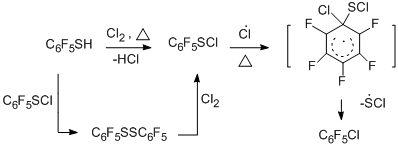
В развитие радикальных термолитических методов получения полифтораренов и –гетаренов предложен новый способ синтеза хлор- и бромполифтораренов и –гетаренов сопиролизом полифторарен- и полифторгетарентиолов с хлором, его источниками или бромом в проточной системе при температурах 300-650°С [46]. При этом происходит замена тиольной группы атомами хлора или брома. Процесс протекает с высокой селективностью, высоким выходом (68-95%) и чистотой (96-99%) получаемых галогенполифторированных соединений. Оптимальная температура для реакций с хлором 400°С, с бромом 500°С.
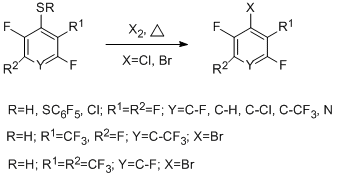
Реакция достаточно гладко протекает как с пентафторбензолтиолом и 4-тетрафторпиридинтиолом (400-500°С), так и с 4-замещёнными 2,3,5,6-тетрафторбензолтиолами, декафтордифенилдисульфидом, 2,4- и 2,5-бис(трифторметил)трифторбензолтиолами, 5-нонафториндантиолом. Схема образования галогенполифтораренов и –гетаренов представлена ниже на примере реакции пентафторбензолтиола с хлором и включает первоначальное превращение тиола в сульфенилхлорид, образование промежуточного радикального σ-комплекса и его трансформацию в конечный продукт.
III. Полифторарены и Zn: реакции гидрогенолиза перфтор- и хлорполифтораренов действием Zn(Cu)-ДМФА-H2O; получение гидрополифтораренов. Полифторароматические цинкорганические соединения
Термолитические реакции различного типа, рассмотренные в разделе II, позволили получить широкий набор перфторированных аренов, -гетаренов и их производных. Однако, как правило, такие реакции в меньшей степени применимы для синтеза не полностью фторированных аренов, поскольку термическая устойчивость последних или их предшественников понижена. Уменьшение термической устойчивости особенно заметно при накоплении в молекуле атомов водорода. Поэтому для получения соединений, содержащих меньшее число атомов фтора, чем их перфторированные аналоги, предложена реакция гидродефторирования последних действием восстановительной системы Zn(Cu)-ДМФА-H2О [47,48]. При действии этого реагента происходит гидрогенолиз С-F связей как в бензильном положении, так и в ароматическом кольце. Добавление NaCl, NH4Cl и других солей, как электролитов, облегчает гидрогенолиз. Без воды реакция гидрогенолиза не идёт.
В серии перфтормоноалкилбензолов, содержащих перфторированные метильную, этильную, пропильную и изопропильную группы, наблюдается высокая селективность [47,49]. Реакция идёт только по С-F связи ароматического кольца в пара- положение к перфторалкильной группе. Предложенный механизм гидрогенолиза Сар –F связи включает образование промежуточного анион-радикала, который распадается на перфторарильный радикал и фторид-ион. Арильный радикал присоединяет электрон, превращаясь в анион, который, взаимодействуя с водой, даёт гидропроизводное [47].
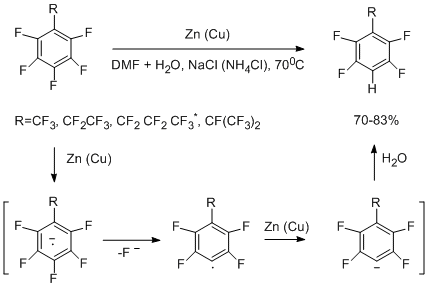
*В отсутствие электролита, DMF - диметилформамид (ДМФА)
В соответствии с квантово-химическими расчётами анион-радикалов октафтортолуола и перфторксилолов [48] реализация наблюдаемого направления реакции связана с локализацией свободного электрона в анион-радикалах в пара-положении к заместителю, что приводит к восстановительному превращению именно в этом положении и получению п-Н-замещённых перфторалкилбензолов. В перфтор-п-ксилоле реакция гидрогенолиза идёт в основном по бензильному положению, причём затрагивается только одна СF3-группа. Содержание 1,4-бис(трифторметил)-3,5,6-трифторбензола в этом случае мало [48].
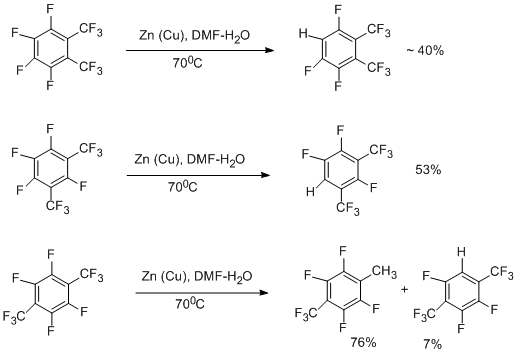
Возможный механизм гидрогенолиза бензильной С-F связи предполагает электронный перенос от поверхности металла на молекулу перфторарена с образованием анион-радикала и его распад.
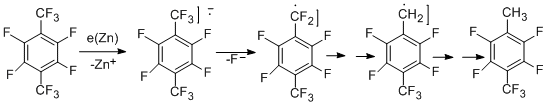
Реакция идёт стадийно с образованием последовательно группировок –CHF2, -CH2F и, наконец, CH3. Выход соединения с СН3-группой может достигать 76%, в то время как содержание других продуктов мало. Перфтор-п-цимол реагирует с участием только перфторизопропильной группы [48].
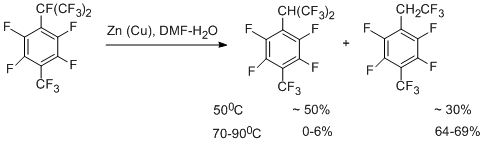
Механизм гидрогенолиза САlk-F связи с образованием гидрированного гексафтор-изопропильного производного аналогичен описанному выше для перфтор-п-ксилола, но здесь есть две возможности распада образующегося анион- радикала – с образованием радикала "А" или "B".
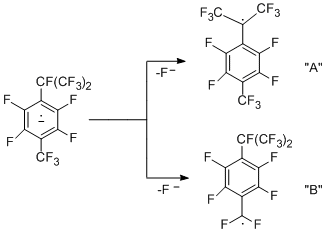
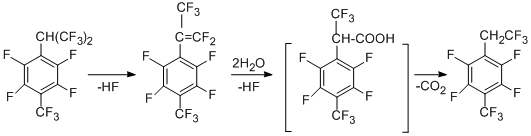
Большая стабильность радикала "А" по сравнению с "B" (согласно квантово-химическим расчётам) и меньшая прочность связи С-F в бензильном положении изопропильной группы может быть одним из основных факторов, определяющих участие в реакции только перфторизопропильной группировки. Соединение с трифторэтильной группой является продуктом дальнейшего превращения гексафторизопропильной группы и получается в водном ДМФА как в присутствии, так и в отсутствие металлов. Его образование можно представить как результат протекающего дегидрофторирования до перфтор-α,4-диметилстирола, который далее реагирует с водой, давая кислоту. Последняя декарбоксилируется и получается 1-трифторметил-2,3,5,6-тетрафтор-4-(2',2',2'-трифторэтил)бензол. Схема подтверждается превращением перфтор-a,4-диметилстирола в данное этильное производное.
Отлично от этого протекает реакция перфтор-4-трет-бутилтолуола с Zn(Cu)-ДМФА-Н2О в присутствии NaCl [49]. В этом случае главным образом идёт дегидрофторирование в орто-положении к объёмной перфтор-трет-бутильной группе.
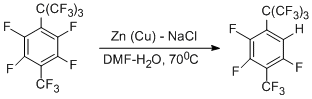
Для некоторых перфтораренов реакция гидродефторирования может протекать в водной среде в присутствии электролитов без добавления ДМФА. В таких условиях, например, в октафтортолуоле и пентафторпиридине также идёт замещение на водород атома фтора в положении 4 [50], хотя при этом остаётся и непрореагировавший исходный арен.
При наличии в алкильной группировке перфторарена атомов хлора или брома реакция гидрогенолиза идёт исключительно с участием этих атомов [51]. Только в случае 4-хлор-2,3,5,6-тетрафторбензотрихлорида помимо продукта основной реакции по бензильному положению (выход 78%) получается 13% продукта гидрогенолиза связи СAr-Сl. Гидрогенолиз связи СAr-Cl вообще является единственным направлением реакции в хлорполифторбензолах (хлорпентафторбензол, смесь изомерных дихлортетрафторбензолов, 1,3,5-трихлортрифторбензол) [51], что связано со структурой образующегося промежуточного анион-радикала σ-типа , в котором электрон взаимодействует с МО, локализованной на связи СAr-Cl.
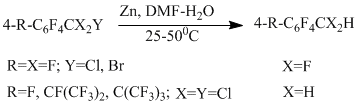
В рассмотренных хлорполифтораренах реакционная способность C-Hal связи соответствует следующим закономерностям: Сбензил-Сl > СAr-Cl; Сбензил-Сl > СAr-F; Сбензил-Сl > Сбензил-F; Сбензил- F < СAr- F (пара-положение).
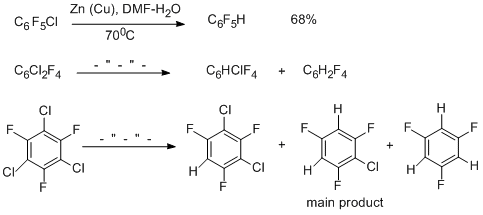
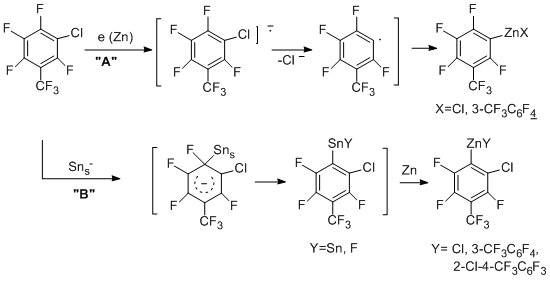
Хлорполифторарены с Zn в ДМФА при нагревании или обработке реакционной массы ультразвуком дают стабильные цинкорганические соединения, которые идентифицируются спектральными методами. Они трансформируются в гидропроизводные при обработке кислотой [51-53]. В реакции участвуют только атомы хлора.
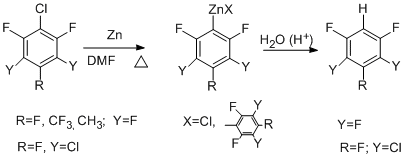
Аналогично реагирует 3-хлоргептафтортолуол. Добавление в систему SnCl2 способствует образованию цинкорганических соединений и за счёт участия в реакции связи СAr–F [54].
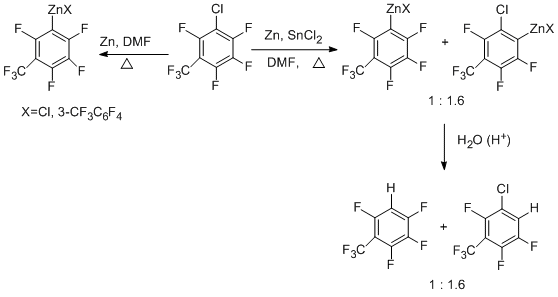
Перфторарены при действии системы Zn-ДМФА-SnCl2 дают цинкорганические соединения с участием связи СAr –F уже при комнатной температуре [55]. Применение ультразвука способствует протеканию реакции. Следует отметить, что ранее прямое образование связи СAr–Zn при участии связи CAr-F в полифтораренах не было отмечено. Найден ряд активности субстратов под действием Zn-SnCl2 и ультразвука (20°С) [55].
Перфториндан образует цинкорганическое производное в положении 5 [53].
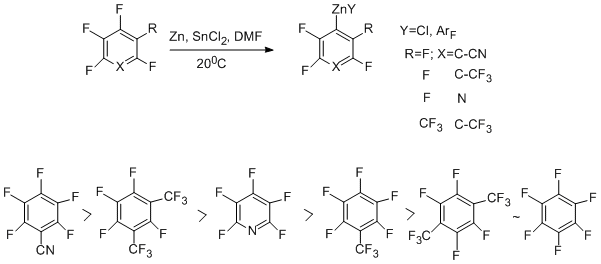
На примере реакции 3-хлоргептафтортолуола представлена схема образования цинкорганических соединений с участием связей С–Cl и CAr –F [54, 56]. В первом случае, по-видимому, электрон поступает на σ-разрыхляющую орбиталь связи C-Cl (путь "А"), в то время как связь С-F затрагивается в результате вклада нуклеофильного механизма (путь "B").
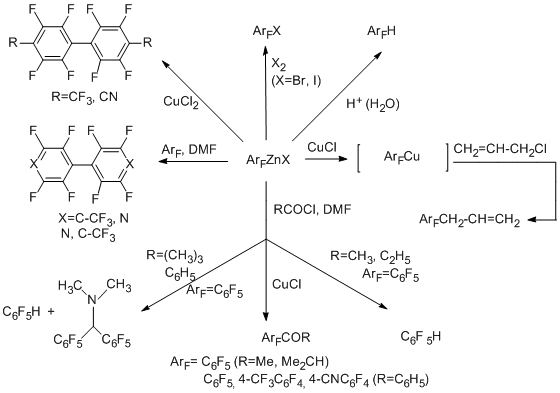
Полученные из перфтораренов цинкорганические производные под действием Н2О (Н+), Вr2, I2 и CuCl2 превращаются соответственно в гидро-, бром-, иодперфторарены и перфторбиарилы [55]. Реакции с аллилхлоридом в присутствии СuCl в качестве катализатора или аллилбромидом дают аллилперфторарены [53]. Реакции с аллилхлоридами в присутствии CuCl, по-видимому, протекают через полифторарилмедьорганические соединения. Полифтордиарилы являются результатом действия на полифторарены полифторарилцинкорганического реагента как нуклеофила. Продукт взаимодействия с хлорангидридами кислот зависит от природы кислоты и условий. Это может быть кетон [57], либо смесь гидроперфторарена и замещённого амина, который получается за счёт участия в процессе реагентов типа Вильсмайера-Хаака. Превращения аналогичного типа реализованы также для цинкфторорганических соединений ряда пиридина и индана.
IV.Химия функциональных производных полифтораренов.
1. Реакция внутримолекулярного нуклеофильного замещения орто-атома фтора в полифтораренах. Синтез гетероциклических и карбоциклических систем
Реакции нуклеофильного ароматического замещения атомов фтора являются наиболее типичным превращением полифтораренов. Замещённые полифторарены при соответствующем строении заместителя и наличии в нём нуклеофильного реакционного центра могут претерпевать реакции внутримолекулярного нуклеофильного замещения с отщеплением орто-атома фтора и образованием гетероциклического или карбоциклического кольца. Это превращение лежит в основе общего и удобного метода получения полифторированных бензгетероциклических соединений, который позволил синтезировать широкий круг самых разнообразных бензгетероциклических систем, в том числе и трудно доступных иным путём. Реакцию можно осуществить, непосредственно действуя на перфторарен бифункциональным нуклеофилом с выделением продукта реакции по одному нуклеофильному центру и последующей циклизацией, или не выделяя первичный продукт реакции с перфторареном. Примеры приведены ниже [58].
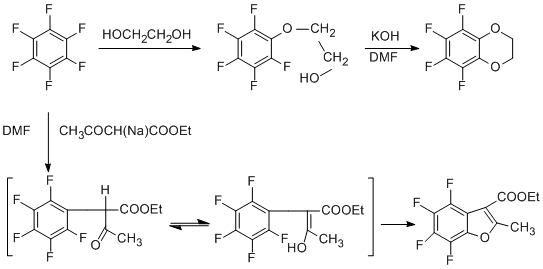
Циклизацией пентафторфенетиламина [59], изомерных трифторфенетиламинов [60] и β-пентафторфенилпропиламина [61] действием KF в ДМФА получены полифторированные производные индолина. Система KF/ДМФА часто используется в реакциях внутримолекулярного нуклеофильного замещения.
Интересные результаты получаются, когда заместитель в полифторарене содержит два нуклеофильных центра. Так, циклизация α-бензамидо-β-пентафторфенилакриловой кислоты идёт только с участием карбоксильной группы
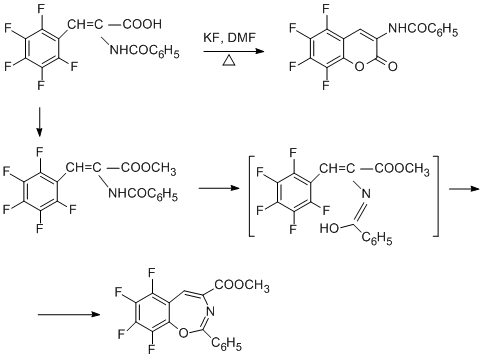
и даёт производное кумарина, в то время как в циклизации метилового эфира этой кислоты участвует бензамидная группа, реагирующая как енол, что приводит к производному бенз[f]оксазепина-1,3 [62]. По-видимому, нуклеофильность бензамидной группы низка, а наличие электроноакцепторного заместителя способствует енолизации этой группы, так что активный нуклеофильный центр оказывается на кислороде.
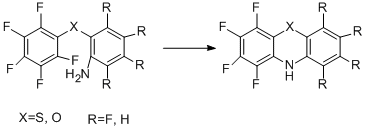
Реакция внутримолекулярного нуклеофильного замещения использовалась и для получения дибензозамещённых полифторированных гетероциклов, например, фенотиазинов [63,64] и феноксазинов [63], а также других систем [65].
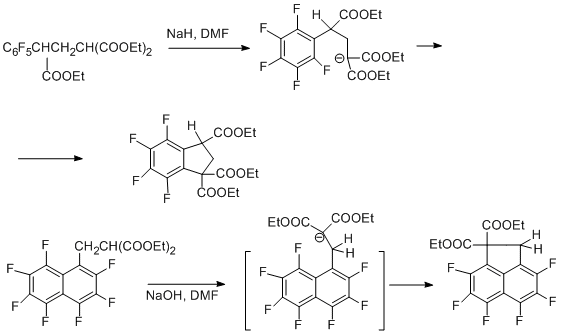
Если нуклеофильный центр находится на углероде, внутримолекулярная циклизация приводит к карбоциклической системе [66].
2. Реакция внутримолекулярной электрофильной циклизации
Описанное выше внутримолекулярное нуклеофильное замещение с отщеплением из орто-положения аниона фтора, как и в целом реакции нуклеофильного замещения, является весьма характерным типом превращений полифтораренов. В то же время внутримолекулярное электрофильное замыкание с отщеплением из орто-положения катиона фтора не является энергетически благоприятным процессом. Однако, как оказалось, внутримолекулярная электрофильная циклизация всё же возможна, что было показано на примерах образования полифторированных карбоциклических соединений. При взаимодействии перфтор-1-фенил- и –3-фенилпропенов с AlCl3 (90-100°C) получается 1,1,3,3-тетрахлоргексафториндан наряду с 1,1,3,3,5-пентахлорпентафторинданом (15%) и небольшим количеством пентафторфенил-2-фтортетрахлорпропена [67].
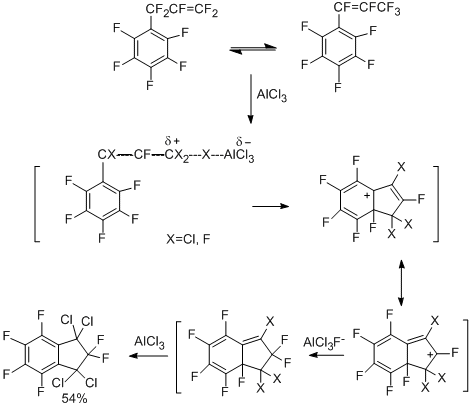
При действии гексафторпропена на пентафторбензол в присутствии AlCl3 также образуются эти соединения, причём первоначально, по-видимому, имеет место электрофильное галогеналкенилирование с замещением атома водорода на пергалогенопропенильную группу и затем образование 1,1,3,3-тетрахлоргексафториндана. Процесс сопровождается обменом фтора на хлор. Схема циклизации может включать активацию 1-пентафторфенил-2-фторполигалогенпропенов под действием AlCl3 с последующим электрофильным орто-присоединением и перемещением галогенов в бициклической системе. Образование 1,1,3,3,5-пентахлорпентафториндана может происходить путём замены атома фтора на хлор в 1,1,3,3-тетрахлоргексафториндане под действием AlCl3 [68].
При взаимодействии перфтор-α, β, о-триметилстирола со SbF5 при 200°С также наблюдалось образование продукта внутримолекулярной электрофильной циклизации – перфтор-1,7-диметилиндана [69].
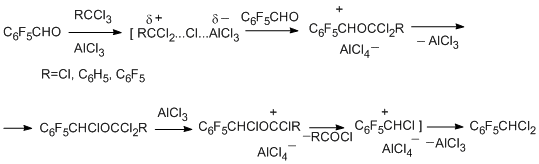
3. Реакция полихлоралкилирования полифтораренов с N-, O- и S-содержащими функциональными группами в присутствии AlCl3, протекающая по гетероатому
Одним из успешно развиваемых направлений трансформации функциональных групп полифтораренов с целью расширения круга полифторароматических производных является реакция электрофильного полихлоралкилирования полифторированных ароматических N-, O- и S-содержащих функциональных производных действием системы RCX3/AlCl3 (X=Cl, F), протекающая с образованием полихлоралкильного фрагмента в a-положении к гетероатому. Метод основан на атаке по донорному N-, O- или S-гетероатому промежуточного электрофильного комплекса, который получается при взаимодействии тригалогенметильного реагента с AlCl3. Образующееся полихлорметильное производное может быть выделено или претерпевает дальнейшие превращения в соединения различных типов. Реакция является достатотчно общей [70,71]. В качестве субстратов использованы полифторированные амины ряда бензола, нафталина, дифенила [72], арилгидразины [73] (нефторированные ароматические амины и гидразины с акцепторной нитрогруппой также вступают в эту реакцию), пентафторфенол [74], пентафторбензальдегид [75], полифторбензолтиолы [76]. Соединения RCX3 включают CCl4 , ароматические производные с одной или несколькими группами CX3, в том числе поли- и перфторированные, а также пергалогенированные олефины с терминальной группой CCl3 (CClX=CYCCl3; X=Cl, C6F5; Y=Cl, F) [77]. Успешному развитию метода способствует замещение в ароматическом кольце субстрата атомов водорода на фтор, что исключает альтернативный путь реакции полихлоралкилирования по связи СAr-H.
Полифторированные ароматические амины [72] и гидразины [73] в реакции с RCX3 и AlCl3 дают N-трихлорметильные производные, которые в условиях реакции превращаются соответственно в N–полифторарилкарбонимидоилдихлориды или N-трихлорметил-N-полифторарилгидразонодихлорметаны, когда R=X=Cl, или в N-полифторарилимидоилхлориды и N-полифторарилгидразонохлориды, когда R=Ar, ArF, а X=Cl, F. Получающиеся карбонимидоилдихлориды и имидоилхлориды идентичны полученным в описанных выше реакциях аминов (раздел II.2.2).
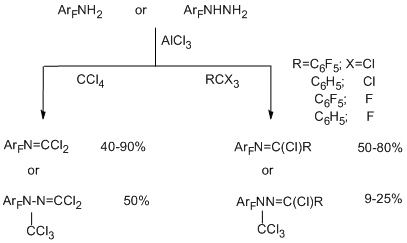
Применение в качестве полихлоралкилирующих реагентов ССl3-замещённых олефинов [77] позволяет получать полигалогенированные 4-аза-1,3-бутадиены.
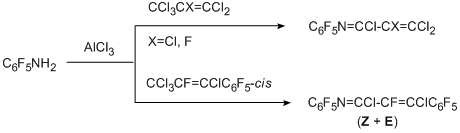
При использовании соединений с несколькими пергалогенированными алкильными группировками (изомерные перфторксилолы, перфтор-п-цимол, 1,3-бис(трихлорметил)-тетрафторбензол, 4-(трихлорметил)ундекафторизопропилбензол) в изученных условиях образование имидоилхлоридной группировки идёт только по одной СХ3-группе и получается большое количество побочных продуктов [78].
Наиболее вероятный путь протекания реакции полигалогеналкилирования представлен на схеме [71]. N-Полихлоралкиламин, образующийся на первой стадии полихлоралкилирования, может быть выделен, если его дальнейшее превращение в имидоилхлоридное производное невозможно, как, например, в случае декафтордифениламина или N-метилпентафторанилина [72].
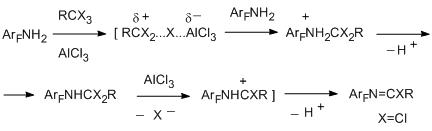
Полихлоралкилирование пентафторфенола действием ССl4 и AlCl3 протекает по аналогичной схеме с образованием первичного продукта полихлоралкилирования – трихлорметилпентафторфенилового эфира, а также бис(пентафторфенил)карбоната, как продукта гидролиза образующегося производного дихлорметана, и трис(пентафторфенокси)хлорметана [74]. Варьированием условий реакции можно cдвинуть систему в сторону образования преимущественно одного из продуктов (например, до 40% первоначального эфира и более 45% карбоната).
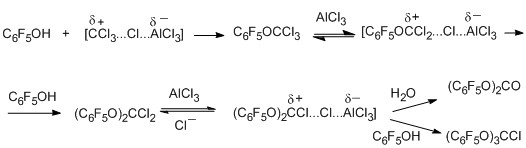
Использование вместо CCl4 бензотригалогенидов ArCX3 (X=Cl, F; Ar=C6H5, C6F5) приводит к получению пентафторфениловых эфиров бензойных кислот – продуктов гидролиза промежуточно образующихся эфиров C6F5OCX2 Ar (выход ~ 50%) [79].
Прямое полихлоралкилирование по атому кислорода пентафторбензальдегида даёт с высоким выходом в качестве конечного продукта пентафторбензилиденхлорид [75].
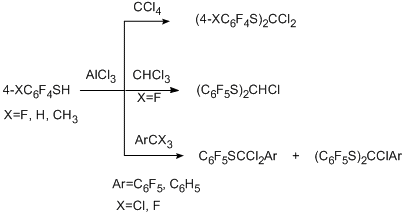
Подобно полифторированным фенолам полифторированные бензолтиолы полихлоралкилируются по атому серы, однако в этом случае первичные продукты полихлоралкилирования получены лишь с бензотригалогенидами, а основными продуктами реакции являются стабильные к гидролизу полифторарилсульфиды с замещённой метиленовой группировкой [76]. Различие в результатах, полученных в реакциях полифторированных фенолов и бензолтиолов, объясняются с позиций более высокой нуклеофильности бензолтиолов и лучшей способности атомов кислорода по сравнению с серой стабилизировать соседний карбокатионный центр.
4. Превращения полифтораренов с имидоилхлоридной группировкой
4.1. Реакции с нуклеофильными реагентами
Полифторароматические имидоилхлоридные производные – высоко реакционноспособные соединения, которые могут участвовать в превращениях различного типа. Наиболее типичными для них являются реакции с нуклеофильными реагентами. Они протекают с участием атомов хлора при N=C связи и сохранением или трансформацией самой этой связи. Продукты реакций либо стабильны, либо претерпевают дальнейшие превращения. Акцепторные свойства полифторароматического кольца и его способность служить дополнительным реакционным центром ведёт к вторичным превращениям, наиболее интересным из которых является гетероциклизация.
4.1.1. N–Нуклеофильные реагенты
Закономерности, которые наблюдаются в реакциях полифторароматических карбонимидоилдихлоридов и имидоилхлоридов с первичными, вторичными и третичными аминами, аналогичны найденным ранее в соответствующих реакциях их нефторированных аналогов. В реакциях карбонимидоилдихлоридов с первичными аминами легко отщепляются оба атома хлора, и тип продукта зависит от природы и количества амина. Реакции с первичными алифатическими аминами дают
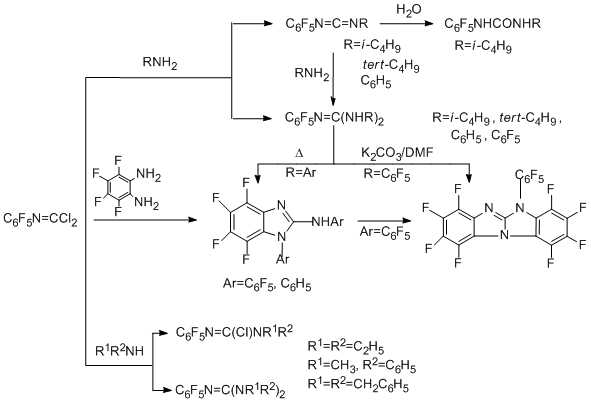
карбодиимиды или гуанидины. С первичными ароматическими аминами образуются только гуанидины. В случае вторичных аминов, независимо от их природы, сначала идёт элиминирование одного атома хлора и образование хлорформамидинов, а затем второго хлора с образованием гуанидинов [80,81]. В реакциях с третичными аминами полифторароматические имидоилхлоридные производные дают имидоиламмониевые соли, которые в случае ароматических аминов (например, 4-N,N-диметиламинопиридина) стабильны и могут быть выделены с количественным выходом [82], в то время как в случае алифатических аминов они образуются как промежуточные соединения, которые превращаются в хлорформамидины [83]. Реакции со всеми аминами протекают по механизму бимолекулярного присоединения- отщепления с образованием на первой стадии промежуточного тетраэдрического интермедиата ТЕ± [83,84]. В случае первичных и вторичных аминов его образование является стадией, лимитирующей скорость, с последующей трансформацией в Z-изомер хлорформамидина (путь "А"). Для третичных аминов лимитирующей скорость стадией является стереомутация первичного тетраэдрического интермедиата ТЕ± в термодинамически более стабильный ТZ± и далее – в соль (путь "B").
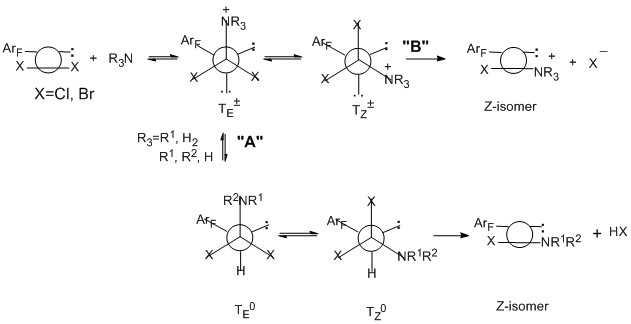
Схема, приведённая ниже, объясняет образование различных продуктов в случае первичных и вторичных аминов (карбодиимиды/гуанидины и хлорформамидины соответственно):
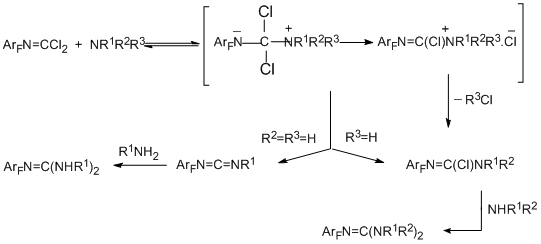
Хлорформамидины и гуанидины получаются также, если проводить реакции полифторарилимидоилдихлоридных производных с ароматическими аминами в присутствии AlCl3 [85]. При действии на N-пентафторфенилкарбонимидоилдихлорид аминов и их полифторированных по кольцу аналогов образование гуанидинов и хлорформамидинов в присутствии AlCl3 идёт в более мягких условиях и с большей степенью превращения. Объясняется это изменением механизма превращений от стадийного бимолекулярнго присоединения-отщепления в отсутствие AlCl3 до механизма, включающего поляризацию имидоилхлоридного производного под действием AlCl3 с образованием положительно заряженного интермедиата, который атакует атом азота амина. Подробнее механизм такого типа будет обсуждён в разделе 4.2.
4.1.2. О- Нуклеофильные реагенты
Наиболее детально исследованы реакции N-пентафторфенилкарбонимидоил-дихлорида со спиртами, фенолами и этиленгликолем [86]. В реакциях без добавления оснований идёт присоединение нуклеофила по связи С=N с образованием в качестве конечных продуктов пентафторфенилкарбаматов. В присутствии основания, например, K2CO3 отщепляется один или оба атома хлора, связь С=N остаётся , и получаются моно- или диимидаты, или, как в случае этиленгликоля, может идти внутримолекулярная гетероциклизация.
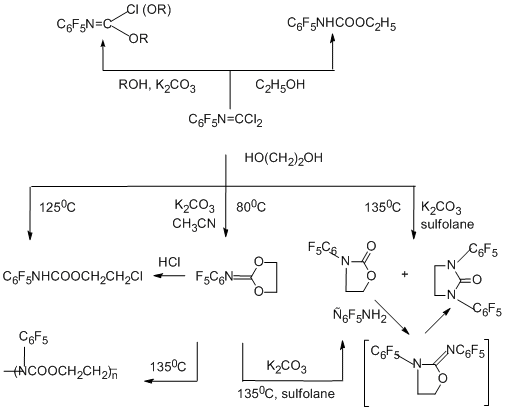
4.1.3. S- Нуклеофильные реагенты
Изучены реакции полифторарилимидоилхлоридных производных с бензолтиолами, тиомочевиной, натрий-N,N-диэтилдитиокарбаматом и источниками SCF3- -аниона [87], но в синтетическом плане интерес представляют лишь три первых реагента.
Основное направление реакций с бензолтиолами в ацетонитриле и присутствии безводного K2CO3 - замещение одного или двух атомов хлора на остаток бензолтиола. При наличии у углеродного атома N=C кратной связи группировки C6F5 наблюдается замещение на нуклеофил пара-атома фтора этой группировки, причём этот процесс превалирует над замещением атома хлора. Связано это с тем, что согласно квантово-химическим расчётам для имидоилхлоридных производных такого типа, например, N-пентафторфенил-(пентафторбенз)имидоилхлорида направление реакции определяется орбитальным контролем, а наибольшие плотности граничной НВМО сосредоточены как раз в пара-положении С-пентафторфенильного кольца и на атоме углерода N=С связи [87]. Для аналога с фенильной группой у С-атома и для N-пентафторфенилкарбонимидоилдихлорида направление реакции определяет выигрыш в энергии образования промежуточного s-комплекса присоединения S-нуклеофила по атому углерода N=C кратной связи.
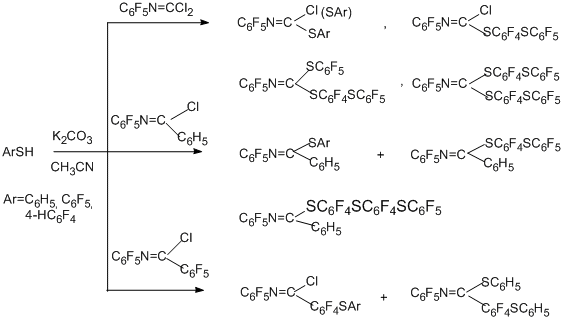
Тиомочевина и натрий-N,N-диэтилдитиокарбамат дают либо пентафторфенилизотиоцианат, либо гетероциклические производные [87].
4.1.4. Галоген-анионы
Стадийное замещение атомов хлора на бром наблюдается в реакции N-пента-фторфенилкарбонимидоилдихлорида при действии LiBr в ацетонитриле или AlBr3 в дибромметане [79].
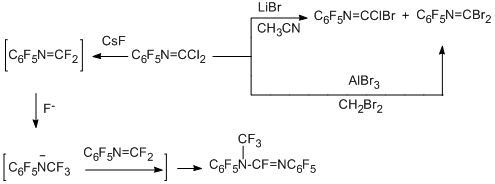
Попытки замещения атомов хлора на фтор и выделения карбонимидоилдифторида действием CsF в различных условиях привели только к получению димера этого дифторида [79].
4.2. Реакции в присутствии AlCl3.
В присутствии AlCl3 полифторарены с имидоилхлоридной группировкой действуют как эффективные электрофилы, что позволяет вводить реакционно-способные ArFN=CCl- или ArFN=CАr-группы в субстраты. В ходе превращений могут идти реакции типа циклоприсоединения или внутримолекулярной циклизации, которые приводят к полифторированным N-гетероциклическим соединениям.
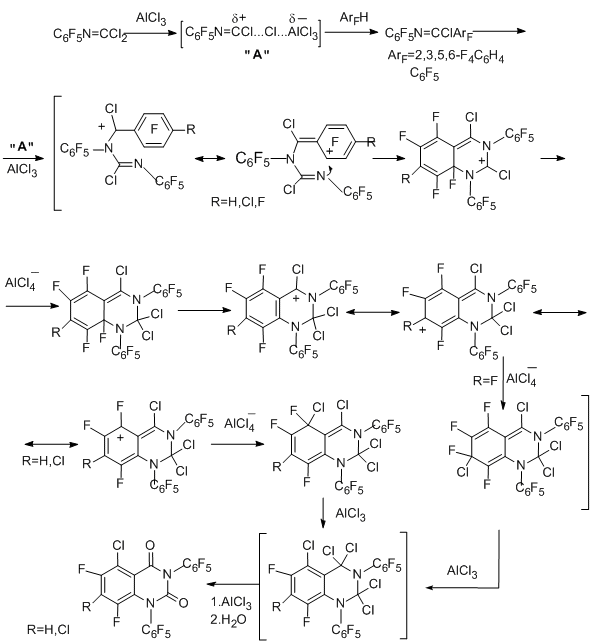
Реакции N- пентафторфенилкарбонимидоилдихлорида с ароматическими углеводородами (бензол, толуол, изомерные ксилолы, мезитилен) идут в мягких условиях, давая стабильные имидоилхлориды, которые далее превращаются в азометины [88]. Наблюдается преимущественная пара-ориентация относительно СН3-группы на обеих стадиях реакции. Реакционная способность углеводородов возрастает в ряду бензол< толуол < п-ксилол < о-ксилол < м-ксилол < мезитилен. Аналогичным образом, но в несколько более жёстких условиях реагируют и менее основные фторбензол и 1,3,5-трифторбензол [89]. Наблюдаемые закономерности в реакциях аренов и фтораренов позволяют заключить, что реакции имеют электрофильный характер и включают поляризацию имидоилхлоридного производного под действием AlCl3 с промежуточным образованием катиона типа нитрилия и последующую атаку углеводорода положительно заряженным центром интермедиата.
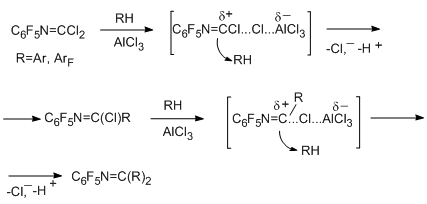
В случае 1,2,4,5-тетрафторбензола и пентафторбензола при повышенной температуре преимущественно идёт внутримолекулярная циклизация, приводящая к полифторированным 1,2,3,4-тетрагидрохиназолин-2,4-дионам. Побочной реакцией является замещение атомов фтора на хлор [89]. Эти данные указывают, что накопление атомов фтора в ароматическом кольце приводит к изменению реакционной способности соединения, и граница наблюдается при переходе от трёх к четырём атомам фтора.
Реакции полифторированных имидоилхлоридных производных с соединениями, содержащими азот-углеродные кратные связи, в присутствии AlCl3 также, по- видимому, протекают с участием катионоидного интермедиата типа нитрилий-катиона, образуя продукты "циклоприсоединения" – пяти- и шестичленные N-гетероциклические соединения. Они могут рассматриваться как новый путь к полифторированным N-гетероциклическим системам.
Так, 4-R-тетрафторфенилкарбонимидоилдихлориды при взаимодействии с N-пентафторфенилтрихлорацетимидоилхлоридом (N=С кратная связь) дают производные 2-имидазолидона, а "димеризация" упомянутого трихлорацетимидоилхлорида в присутствии AlCl3 даёт замещённый 2-имидазолон [90]. Различие в продуктах реакции "димеризации" и взаимодействия двух разных имидоилхлоридных производных можно объяснить с позиций большей стабильности и лёгкости образования катионоидных интермедиатов, когда Х=Сl по сравнению с Х=ССl3.
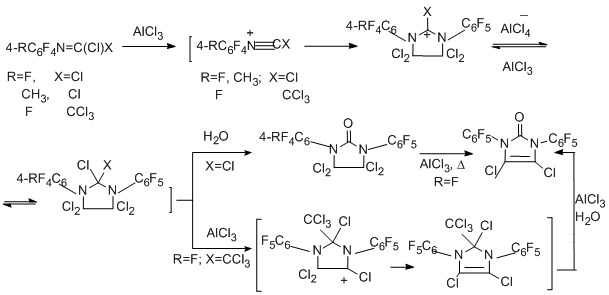
Замещённый триазин был получен в реакции с бензонитрилом (N≡C кратная связь) при высокой температуре [90].
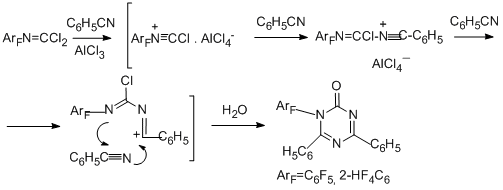
Наконец, взаимодействие N- пентафторфенилкарбонимидоилдихлорида с бензойной и фталевой кислотами или их производными (хлорангидридами, этилбензоатом, фталевым ангидридом) при 170°С в присутствии AlCl3 приводит в качестве основного продукта к 2-(пентафторфенил)изоиндолин-1,3-диону, причём в случае фталевой кислоты и её производных выход продукта составляет более 90% [91].
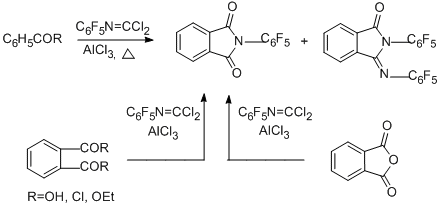
2-(Пентафторфенил)-3-[(пентафторфенил)имино]изоиндолин-1-он является вторым основным продуктом в реакции с хлористым бензоилом. В случае бензойной кислоты основной продукт – бензоильное производное пентафторанилина. Возможные пути образования наблюдаемых соединений в случае реакции с хлористым бензоилом представлены на схеме ниже.
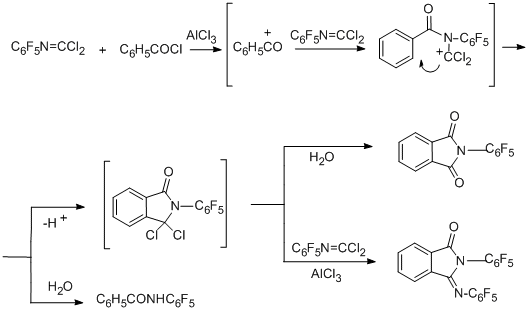
О реакциях полифторароматических имидоилхлоридных производных с ароматическими аминами в присутствии AlCl3 см. раздел IV.4.1.1.
5. Нитрование и нитрозирование полифторароматических N-алкиламинов. Получение N-нитро- и N-нитрозо- N-алкилполифторариламинов
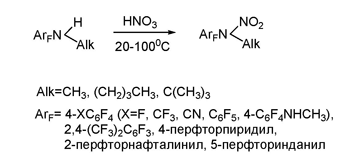
Развитием методов трансформации функциональных групп полифтораренов под действием электрофильных реагентов является нитрование и нитрозирование полифторированных N-алкиламинов.
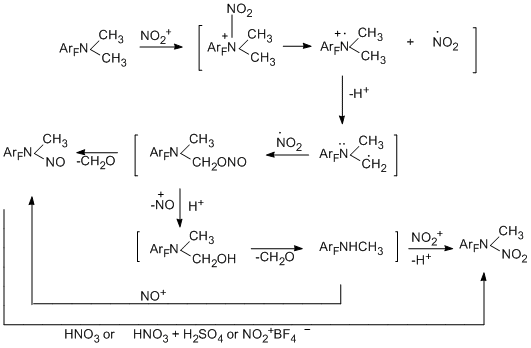
Взаимодействие N-алкилполифторариламинов с азотной кислотой приводит к N-нитро-N-алкилпроизводным [92]. В некоторых случаях в качестве минорных продуктов получаются N-нитрозосоединения. Реакция является достаточно общей и удовлетворительно протекает даже при понижении основности амина за счёт введения дополнительных акцепторных группировок или использовании достаточно объёмных алкильных групп. В случае трет-бутильной группы N-нитроамины мало стабильны, и об их образовании можно судить только по спектральным данным.
Механизм нитрования включает электрофильную атаку атома азота аминогруппы нитроний-катионом, генерирующимся из азотной кислоты. Участие в реакции нитроний-катиона подтверждается образованием тех же N-нитроалкиламинов при использовании заведомого источника такого катиона – тетрафторбората нитрония в сульфолане. Правда, в этом случае в качестве минорного продукта получаются N-нитрозосоединения.
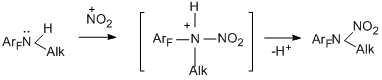
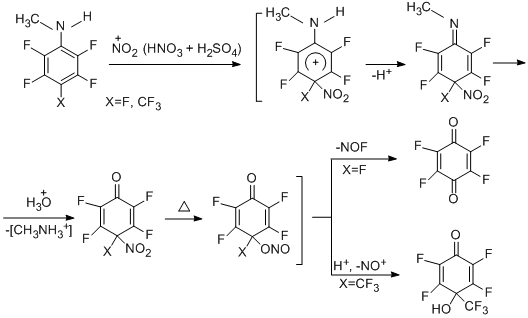
На примере реакций N-метилпентафторанилина и N-метилперфтор-п-толуидина со смесью азотной и серной кислоты показана возможность образования продуктов иного типа – тетрафтор-п-бензохинона и гептафтор-п-толухинола соответственно, что может быть связано с уменьшением основности атома азота аминогруппы за счёт протонирования и, как следствие, атаки нитроний-катионом по атому углерода ароматического кольца [92].
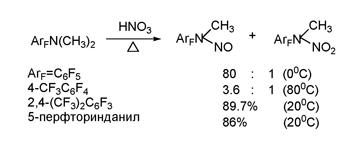
Реакции полифторароматических N,N-диметиламинов с азотной кислотой и со смесью HNO3+H2SO4 идут с конверсией боковой цепи, и главными продуктами этих превращений являются N-нитрозопроизводные в виде смеси Е и Z-изомеров с преобладанием первых (~2.2-3:1) [93].
N-Нитрозопроизводные под действием смеси HNO3 и H2SO4, HNO3 или NO2BF4 превращаются в соответствующие N-нитропроизводные [93]. Схема ниже представляет возможные пути образования N-нитрозо- и N-нитроалкиламинов [93].
N-Нитрозо- N-алкилполифторариламины, которые могут представить интерес как биологическиактивные соединения, можно получать с выходом от 56 до 94% при нитрозировании N-моноалкилполифторариламинов действием NaNO2 в HCl или CH3COOH [94]. При этом образуется смесь E и Z-изомеров, соотношение которых зависит от природы исходного амина.
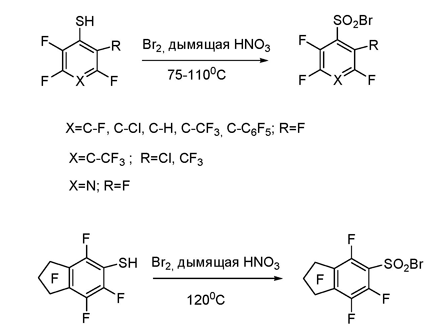
6 Реакции полифторарилтиолов с электрофилами. Синтез полифторарилсульфонилбромидов
Другим примером электрофильной трансформации функциональной группы полифторарена или -гетарена является превращение тиольной группировки полифтортиолов ряда бензола, бифенила, индана и пиридина в сульфонилбромидную под действием Вr2 – дымящая HNO3 или других окислительных систем (Br2 + HNO3 +H2SO4 , НВr + HNO3 + H2SO4 и др.). В большинстве случаев достигается высокий выход сульфонилбромида (70-91%). В некоторых реакциях пентафторбензолтиола было отмечено образование небольшого количества побочного продукта – декафтордифенилдисульфида- , в частности, при использовании систем Br2 + H2SO4, Br2 +CH3COOH, Br2 + H2O. На примере этого соединения было показано, что под действием Br2 и дымящей HNO3 дисульфиды также превращаются в соответствующие сульфонилбромиды [95]. Таким образом, предложен достаточно простой и удобный метод трансформации тиольной группы в сульфонилбромидную, способную к дальнейшим превращениям, в том числе и в соединения с практически полезными свойствами.
Предполагается, что в образовании сульфонилбромидной группировки участвуют электрофильные бромирующие агенты и нитроний-катион. Один из возможных маршрутов протекания реакции может включать образование интермедиата ArFSBr за счёт взаимодействия полифторарентиола с электрофильными агентами (этот же интермедиат может отвечать и за образование ArFS-SАrF). Дальнейшее взаимодействие интермедиата ArFSR (R=Br, SArF) c Br2 и ONO2- даёт другой интермедиат ArFS(O)Br, который далее превращается в конечный продукт.
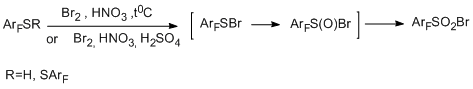
VI. Заключение
Рассмотренные реакции полифторароматических соединений продемонстрировали новые синтетические возможности в этой области химии, а также привели к исследованию ряда аспектов фундаментального характера. Выяснение новых закономерностей реакционной способности полифторароматических соединений будет способствовать развитию их синтетических возможностей. Исследование может оказаться весьма полезным в целом для органической химии. Найденные новые синтетические возможности полифтораренов могут стимулировать интерес к поиску новых областей их практического использования.
Литература
- Ворожцов-мл. Н.Н., Платонов В.Е., Якобсон Г.Г.//Изв.АН СССР. Сер. хим. 1963.N. 8. С.1524
- Ворожцов-мл.H.Н., Якобсон Г.Г. // Журн. общ. химии.1957. Т.27.Вып. 6. С.1672-1676.
- Ворожцов-мл. Н.Н., Якобсон Г.Г. // Журн. общ. химии.1961. Т.31. Вып. 11. С.3705-3708.
- Якобсон Г.Г., Платонов В.Е.,Ворожцов-мл. Н.Н. // Журн. общ. химии.1965.Т.35.Вып. 7.С 1158-1161.
- Якобсон Г.Г., Штейнгарц В.Д., Ворожцов-мл. Н.Н. // Изв. АН СССР. Сер. хим.1964.N 8.С.1551.
- Якобсон Г.Г., Власов В.М., Ворожцов-мл.Н.Н. // ДАН СССР. 1966.Т.169.N. 4. С.855-857.
- Якобсон Г.Г., Одиноков В.Н., Петрова Т.Д., Ворожцов-мл. Н.Н. // Журн. общ.химии.1964.Т.34.Вып.9.С.2953-2958.
- Якобсон Г.Г., Одиноков В.Н., Ворожцов-мл. Н.Н. // Журн. общ. химии.1966.Т.36.Вып. 1.С. 139-142.
- Власов В.М., Одиноков В.Н., Рудакова Р.И., Якобсон Г.Г., Ворожцов-мл.Н.Н. //Журн. общ. химии. 1967. Т. 37. Вып. 1. С. 171-176.
- Якобсон Г.Г., Штейнгарц В.Д., Костина Н.Г., Осина О.И., Ворожцов-мл.Н.Н. //Журн. общ. химии. 1966.Т.36. Вып. 1. С. 142-147.
- Якобсон Г.Г., Платонов В.Е., КрюковаВ.С., Герштейн Н.А., Ворожцов-мл.Н.Н.Журн. общ. химии. 1966. Т. 36. Вып. 12. С. 2131-2135.
- Yakobson G.G., Petrova T.D., Kobrina L.S. // Fluorine Chem. Rev. 1974.V.7.P.115-223.
- Фурин Г.Г., Юматов В.Д. Новое в химии полифторароматических соединений.Новосибирск : Изд-во НГПУ. 2006. 218 с.
- Ворожцов-мл. Н.Н.,Езерский С.Н., Коллегов В.Ф., Львова А.Я., Платонов В.Е.,Пушкина Л.М., Соколов С.В., Татауров Г.П., Якобсон Г.Г.//Изв. АН СССР. Сер.хим. 1966.N. 7.С. 1291.
- Ворожцов-мл. Н.Н., Дворникова К.В., Коллегов В.Ф., Платонов В.Е.,. ПушкинаЛ.М., Соколов С.В., Татауров Г.П.,Якобсон Г.Г.//Журн. ВХО им. Д.И.Менделеева. 1969. Т. 14.N. 1.С. 114.
- Платонов В.Е., Ермоленко Н.В., Якобсон Г.Г., Ворожцов-мл. Н.Н.//Изв. АНСССР. Сер.хим. 1968.N 12.С. 2752-2754.
- Ворожцов-млН.Н.,ЕрмоленкоН.В.,МазаловС.А.,ОсинаО.И.,ПлатоновВ.Е.,ТюринВ.С.,ЯкобсонГ.Г.//Изв.АН СССР. Сер. хим. 1969.N 1.С. 196.
- Платонов В.Е., Ермоленко Н.В., Якобсон Г.Г. // Изв. СО АН СССР. Сер. хим.наук.1978. Вып. 2. N. 4. С. 117-123.
- Платонов В.Е. Дисс. докт. хим. наук, НИОХ СО РАН.Новосибирск. 1979.
- Платонов В.Е., Ермоленко Н.В., Якобсон Г.Г. // Изв.АН СССР. Сер. хим. 1970.N12. С. 2843.
- Платонов В.Е., Малышева В.В., Якобсон Г.Г. // Изв. СО АН СССР. Сер. хим.наук.1977.Вып. 2. N. 4. С.133-141.
- Якобсон Г.Г., Платонов В.Е., Ермоленко Н.В., Коллегов В.Ф., Черток В.С. // Изв.СО АН СССР. Сер. хим. наук. 1971. Вып. 1. N. 2. С.100-102.
- Осина О.И., Черток В.С., Платонов В.Е., Якобсон Г.Г. // Изв. СОАН СССР, Сер.хим. наук.1978. Вып. 6. N. 14. С. 138-141.
- Петрова Т.Д.,Платонов В.Е., Горфинкель М.И., Якобсон Г.Г. // Журн. орган.химии.1975. Т. 11. N. 10. С. 2123-2129.
- Платонов В.Е., Фурин Г.Г., Малюта Н.Г., Якобсон Г.Г. // Журн. орган. химии.1972. Т. 8. N. 2. С. 430-431.
- Malyuta N.G., Platonov V.E., Furin G.G., Yakobson G.G. // Tetrahedron. 1975. V. 31.N. 9. P. 1201-1207.
- Платонов В.Е., Гатилова В.П., Дворникова К.В., Якобсон Г.Г.//Изв. АН СССР.Сер. хим. 1974.N. 7.С. 1668-1669.
- ЯкобсонГ.Г.,ПлатоновВ.Е.,ФуринГ.Г.,МалютаН.Г.,ЕрмоленкоН.В.//Изв.АН СССР. Сер. хим. 1971.N. 11.С. 2615.
- Platonov V.E., Yakobson G.G. // Synthesis. 1976. N. 6. P. 374-384.
- Максимов А.М., Моторина Е.Е., Платонов В.Е., Якобсон Г.Г.//IIIВсесоюзнаяконференция по химии фторорганическихсоединений, Одесса (Россия). 1978.Тезисы докладов, С.73.
- Платонов В.Е., Максимов А.М., Якобсон Г.Г.//Изв. АН СССР. Сер. хим. 1977.N.10.С. 2387-2388.
- Максимов А.М., Платонов В.Е., Якобсон Г.Г., Дерягина Е.Н., Воронков М.Г. //Журн. орган. химии.1979.Т.15. Вып. 9. С. 1839-1843.
- Иванова Е.П., Карпов В.М., Платонов В.Е., Татауров Г.П., Якобсон Г.Г.,Яхлакова О.М.//Изв. АН СССР. Сер.хим. 1972.N. 3.С. 733.
- МаксимовА.М.,ПлатоновВ.Е.,ЯкобсонГ.Г.,ЯхлаковаО.М.,БеккерР.А.,ДяткинБ.Л.,КнунянцИ.Л.//Изв.СО АН СССР. Сер. хим. наук. 1981. Вып. 6.N.6.С. 128-133.
- Platonov V.E., Yakobson G.G. // Soviet Scientific Revievs. Section B. ChemistryReviews. Ed. Vol’pin M.E. Amsterdam: Harwood Acad. Publishers. 1984. V. 5. P. 297-345.
- Дворникова К.В., Платонов В.Е., Якобсон Г.Г.//Журн. орган. химии.1975. Т. 11.Вып. 11.С. 2383-2387.
- Дворникова К.В., Платонов В.Е., Якобсон Г.Г.//Изв. АН СССР. Сер. хим. 1978.N. 5.С. 1223.
- Dvornikova K.V., Platonov V.E., Yakobson G.G. // J. Fluorine Chem. 1985. V. 28. N.1-2. P. 99-113.
- Savchenko T.I., Petrova T.D., Platonov V.E., Yakobson G.G. // J. Fluorine Chem.1977. V. 9. N. 6. P. 505-508.
- Савченко Т.И., Петрова Т.Д., Платонов В.Е., Якобсон Г.Г. // Журн. орган. химии.1979. Т. 15. Вып. 5. С. 1018-1024.
- СавченкоТ.И., Петрова Т.Д., Платонов В.Е., Якобсон Г.Г. // Журн. орган. химии.1979. Т. 15. Вып. 5. С. 1025-1029.
- Maksimov A.M., Platonov V.E. // Heteroatom. Chem. 1992. V. 3. N. 4. P. 373-384.
- Platonov V.E., Maksimov A.M., Maslovsky P.I. // J. Fluorine Chem. 1995. V. 75. Is. 1.P. 41-49.
- Platonov V.E.,OsinaО.I.,Maksimov A.M., Kolechkina V.G. // J. Fluorine Chem.1999. V. 96. N. 2. P. 191-192.
- Колечкина В.Г., Максимов А.М., Платонов В.Е., Осина О.И.//Изв. АН. Сер. хим.2001.N. 2.С. 307-309.
- Платонов В.Е.,Максимов А.М., Дворникова К.В., Никульшин П.В.//Журн.орган. химии. 2005. Т. 41. Вып. 11. С. 1681-1687.
- Краснов В.И., Платонов В.Е.//Журн. орган. химии. 1993. Т. 29. Вып. 5. С. 1078-1079.
- Krasnov V.I., Platonov V.E., Beregovaya I.V., Shchegoleva L.N.// Tetrahedron. 1997.V. 53. Is. 5. P. 1797-1812.
- Краснов В.И., Платонов В.Е.//Журн. орган. химии. 2001. Т. 37. Вып. 4. С. 552-557.
- Краснов В.И. Дисс. канд. хим. наук.НИОХ СО РАН. Новосибирск. 1999.
- Krasnov V.I., Platonov V.E. //J. Fluorine Chem. 1992.V. 58. N. 2-3. P. 246.
- Краснов В.И., Платонов В.Е.//Журн. орган. химии. 2000. Т. 36. Вып. 10. С. 1524-1534.
- Виноградов А.С., Краснов В.И., Платонов В.Е.//Журн. орган. химии. 2008. Т. 44.Вып. 1. С. 101-107.
- Krasnov V.I., Vinogradov A.S., Platonov V.E.// Mendeleev Commun. 2006. N.3. P.168-170.
- Miller A.O., Krasnov V.I., Peters D., Platonov V.E., Miethchen R. // Tetrahedron Lett.2000. V. 41. P. 3817-3819.
- Platonov V.E. // 15thEuropean Symposium on Fluorine Chemistry, Prague (CzechRepublic). 2007. Book of Abstrs. Р. 157.
- Vinogradov A.S., Krasnov V.I., Platonov V.E. // Mendeleev Commun. 2008. V. 18.N.4. P. 227-228.
- Якобсон Г.Г., Петрова Т.Д., Канн Л.И., Савченко Т.И., Петров А.К., Ворожцов-мл.Н.Н. // ДАН СССР. 1964. Т. 158. N. 4. С. 926-928.
- Петров В.П., Бархаш В.А., Щеголева Г.С., Петрова Т.Д., Савченко Т.И., ЯкобсонГ.Г. // ДАН СССР. 1968.Т.178. N. 4. С. 864-867.
- Петрова Т.Д., Савченко Т.И., Куковинец О.С., Якобсон Г Г. //Изв. СОАН СССР.Сер. хим. наук. 1974. Вып. 2.С. 117-123.
- Петрова Т.Д., Савченко Т.И., Ардюкова Т.Ф., Якобсон Г.Г. //Изв. СОАН СССР.Сер. хим. наук. 1970. Вып.3.N. 7.С.119-122.
- Петрова Т.Д. , Мамаев В.П., Якобсон Г.Г., Ворожцов-мл. Н.Н. // Химия гет. соед.1968. N. 5. С. 771-776.
- Якобсон Г.Г., Фурин Г.Г., Кобрина Л.С., Ворожцов-мл. Н.Н. // Журн. общ. химии.1967. Т. 37. N. 6. С. 1285-1289.
- Якобсон Г.Г., Фурин Г.Г., Кобрина Л.С., Ворожцов-мл. Н.Н. // Журн. общ. химии.1967. Т. 37. N. 6. С. 1289-1293.
- Brooke G.М. // J. Fluorine Chem. 1997. V. 86. Is. 1. P. 1-76.
- Vlasov V.M., Petrova T.D., Yakobson G.G. // J. Fluorine Chem. 1972/1973. V. 2. N. 4.P. 373-380.
- Платонов В.Е., Дворникова К.В.//Изв. АН СССР. Сер. хим. 1989.N11.С. 2854-2857.
- Карпов В.М., Платонов В.Е., Чуйков И.П., Якобсон Г.Г. // Журн. орган. химии.1983. Т. 19. Вып. 10. С. 2164-2173.
- Карпов В.М., Меженкова Т.В., Платонов В.Е. //Изв. АН СССР. Сер. хим. 1990.N3.С 645-652.
- Петрова Т.Д., Платонов В.Е.//Химия в интересах устойч. развит. 2007. Т. 15.N.5.С. 511-523.
- Petrova T.D., Platonov V.E. // J. Fluorine Chem. 2005. V. 126. Is. 6. P. 860-876.
- Savchenko T.I., Kolesnikova I.V., Petrova T.D., Platonov V.E. // J. Fluorine Chem.1983. V. 22 N. 5. P. 439-458.
- Петрова Т.Д.,РябичевА.Г.,Савченко Т.И., Платонов В.Е., Маматюк В.И.,Гатилов Ю.В., Багрянская И.Ю. // Журн. орган. химии. 1986. Т. 22.Вып. 6.С.1297-1306.
- Петрова Т.Д.,Рябичев А.Г.,Савченко Т.И., Колесникова И.В., Платонов В.Е.//Журн. орган. химии.1988. Т. 24.Вып. 7. С.1513-1517.
- Петрова Т.Д., Платонов В.Е.,Покровский Л.М.//Изв. АН. Сер. хим., 2002.N 3.С.504-506.
- Petrova T.D., Platonov V.E., Maksimov A.M. //J. Fluorine Chem. 1999. V. 98. N. 1. P.17-28.
- Колесникова И.В.,Рябичев А.Г.,Петрова Т.Д., Платонов В.Е. //Изв. АНCCCР.Сер. хим. 1988.N 7.С. 1651-1654.
- Петрова Т.Д., Попова И.С., Колесникова И.В.,Платонов В.Е. //Изв. АНCCCР.Сер. хим. 1994.N. 6.С. 1089-1094.
- Петрова Т.Д.Дисс. докт. хим. наук.НИОХ СО РАН. Новосибирск. 1995.
- Колесникова И.В., Петрова Т.Д.,Платонов В.Е., Рябичева Т.Г., Михайлов В.А.,Попов А.А., Савѐлова В.А. // Журн. орган. химии.1989. Т. 25. Вып. 8. С. 1689-1695.
- Kolesnikova I.V., Petrova T.D., Platonov V.E., Mikhailov V.A., Popov A.A., SavelovaV.A. // J. Fluorine Chem. 1988. V. 40. N 2-3. P. 217-246.
- Крючкова Е.Н., Колесникова И.В., Дрижд Л.П., Савѐлова В.А., Петрова Т.Д.,Платонов В.Е. // Журн. орган. химии.1988. Т. 24.Вып. 10.С. 2042-2046.
- Михайлов В.А., Савѐлова В.А., Попов А.А., Петрова Т.Д.,Платонов В.Е. // Журн.орган. химии.2002. Т. 39. Вып. 8. С. 1187-1195.
- Савѐлова В.А., Попов А.А., Петрова Т.Д., Платонов В.Е., Михайлов В.А. //Теорет. и эксп. химия.2003. Т. 39. N 1. С. 22-26.
- Петрова Т.Д., Платонов В.Е. // Журн. орган. химии.1997. Т. 33.Вып. 5.С. 745-749.
- Петрова Т.Д., Колесникова И.В., Савченко Т.И., Платонов В.Е. // Журн. орган.химии.1984. Т. 20. Вып. 6. С. 1197-1204.
- Petrova T.D., Platonov V.E., Shchegoleva L.N., Maksimov A.M., Haas A., SchelvisM., Lieb M. // J. Fluorine Chem. 1996. V. 79. N.1. P. 13-25.
- Петрова Т.Д., Колесникова И.В.,Маматюк В.И., Ветчинов В.П., Платонов В.Е.,Багрянская И.Ю., Гатилов Ю.В. //Изв. АН. Сер. хим. 1993.N. 9.С. 1605-1611.
- Petrova T.D., Platonov V.E., Pokrovskii L.M., Rybalova T.V., Gatilov Yu.V. // Collect.Czech. Chem. Commun. 2002. V. 67. N. 10. P. 1449-1466.
- Petrova T.D., Platonov V.E., KolesnikovaI.V., Rybalova T.V., Bagryanskaya I.Yu.,Gatilov Yu.V. // J. Fluorine Chem. 2000. V. 103. N. 1. P. 63-73.
- Петрова Т.Д., Платонов В.Е. Журн. орган. химии.2005. Т. 41.Вып. 2.С. 228-236.
- Platonov V.E., Haas A., Schelvis M., Lieb M., Dvornikova K.V., Osina O.I., GatilovYu.V. //J. Fluorine Chem. 2001. V. 109. Is. 2. P. 131-139.
- Platonov V.E., Haas A., Schelvis M., Lieb M., Dvornikova K.V, Osina O.I. //J.Fluorine Chem. 2002. V. 116. Is. 1. P. 3-8.
- Platonov V.E., Haas A., Schelvis M., Lieb M., Dvornikova K.V., Osina O.I., RybalovaT.V., Gatilov Yu.V. // J. Fluorine Chem. 2002. V. 114. Is. 1. P. 55-61.
- Platonov V.E., Bredikhin R.A., Maksimov A.M., Kireenkov V.V. // J. Fluorine Chem.2010. V. 131. Is. 1. P. 13-16.
Материал рекомендован к публикации членом редколлегии В.Е. Платоновым
Fluorine Notes, 2011, 77, 1-2
